(Video 37.1). These include six or more café au lait spots (Fig. 37.1), two or more neurofibromas, café au lait freckles in the axillary or inguinal regions (Crowe sign) (Fig. 37.2), optic nerve glioma, two or more Lisch nodules (pigmented iris hamartomas), sphenoid bone dysplasia or thinning of long bone cortex, and a first-degree relative with NF1. Due to the increased incidence of CNS tumors and optic gliomas, periodic imaging of the brain and orbits is recommended.
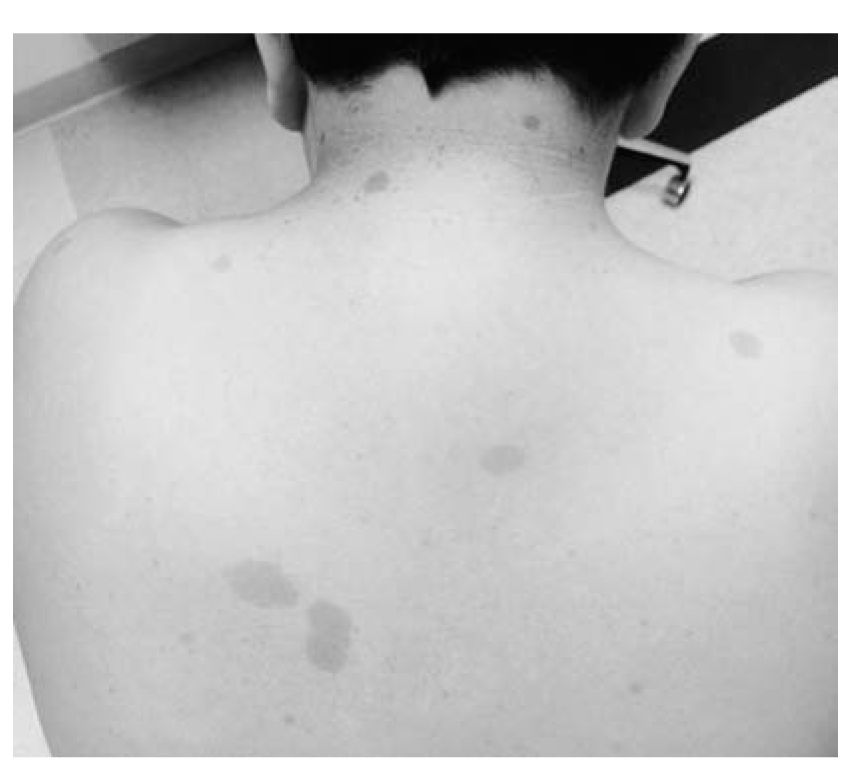
FIGURE 37.1 Café-au-lait spots in a child with NF1. (See color plates.)
Neurofibromatosis type 2 (NF2) is much less common and clinical manifestations arise predominantly in early adulthood. The hallmark lesion of NF2 is bilateral vestibular schwannomas (BVS). These typically occur in early adulthood, but are occasionally seen in adolescence or even childhood (Fig. 37.3). BVS eventually cause progressive hearing loss, tinnitus, and loss of balance. Schwannomas of the trigeminal nerve, spine, and peripheral nerves as well as other CNS tumors have been reported in adults. Eye findings include cataracts and retinal hamartomas.
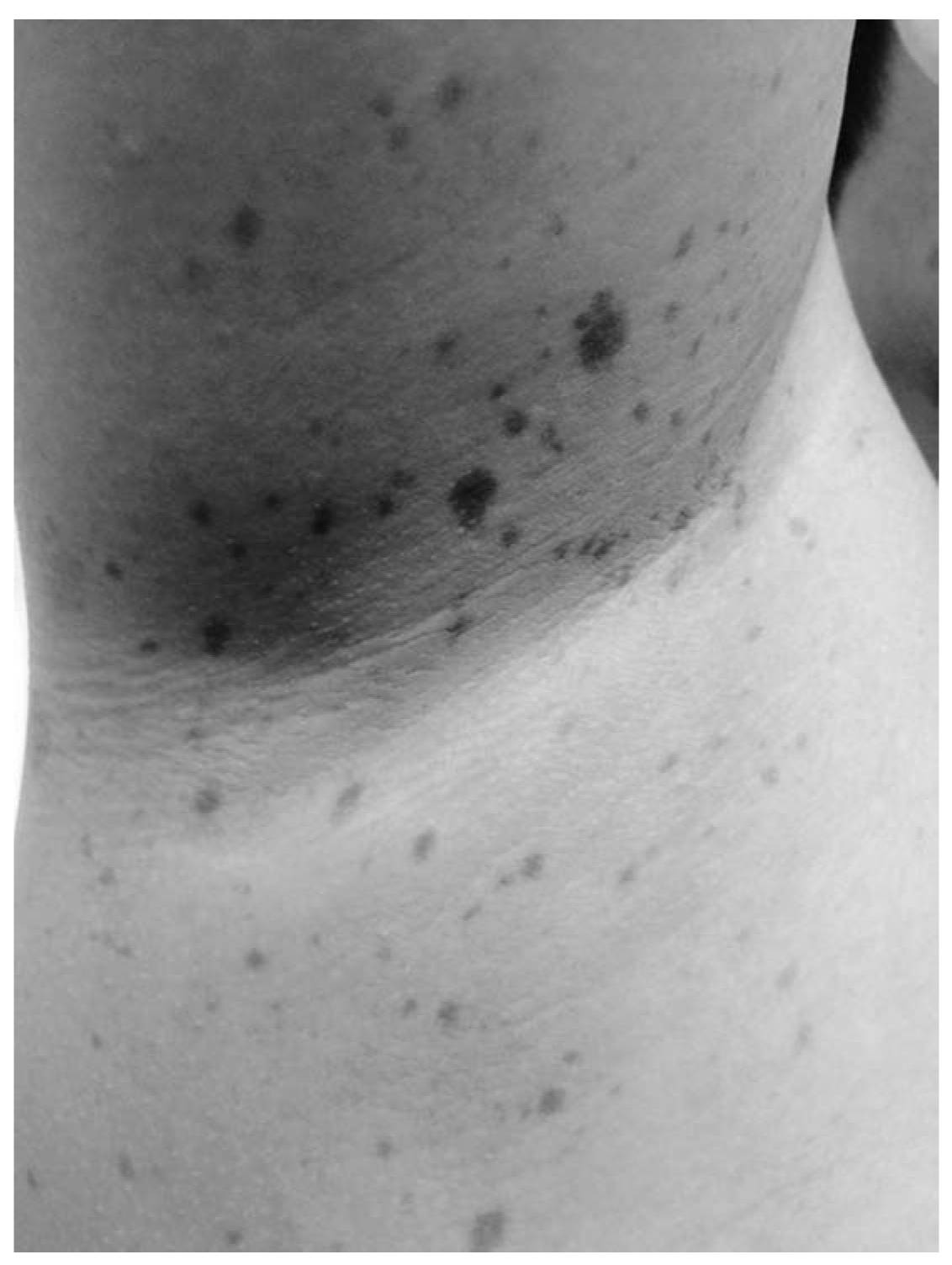
FIGURE 37.2 Axillary freckling in a child with NF1. (See color plates.)
TSC is an autosomal-dominant disorder that affects the brain, skin, kidneys, and heart. Mutations have been localized to the TSC1 gene on chromosome 9 and the TSC2 gene on chromosome 16. These genes code for the proteins hamartin and tuberin. TSC has several characteristic skin lesions, including hypopigmented macules (Fig. 37.4), shagreen patches (Fig. 37.5), adnoma sebaceum, and subungual fibromas. Cortical hamartomas (tubers), subependymal nodules, and subependymal giant cell astrocytomas are seen in the brain. Cardiac rhabdomyomas and renal angiomyolipomas also occur. Children with TSC are at risk for infantile spasms, partial seizures, ASD, and GDD.
Sturge–Weber syndrome presents with port wine birthmarks in the trigeminal nerve distribution and ipsilateral brain hemangiomas. Affected children also have contralateral focal motor seizures and hemiparesis. Other rare neurocutaneous syndromes associated with developmental delays and epilepsy are incontinentia pigmenti and hypomelanosis of Ito.
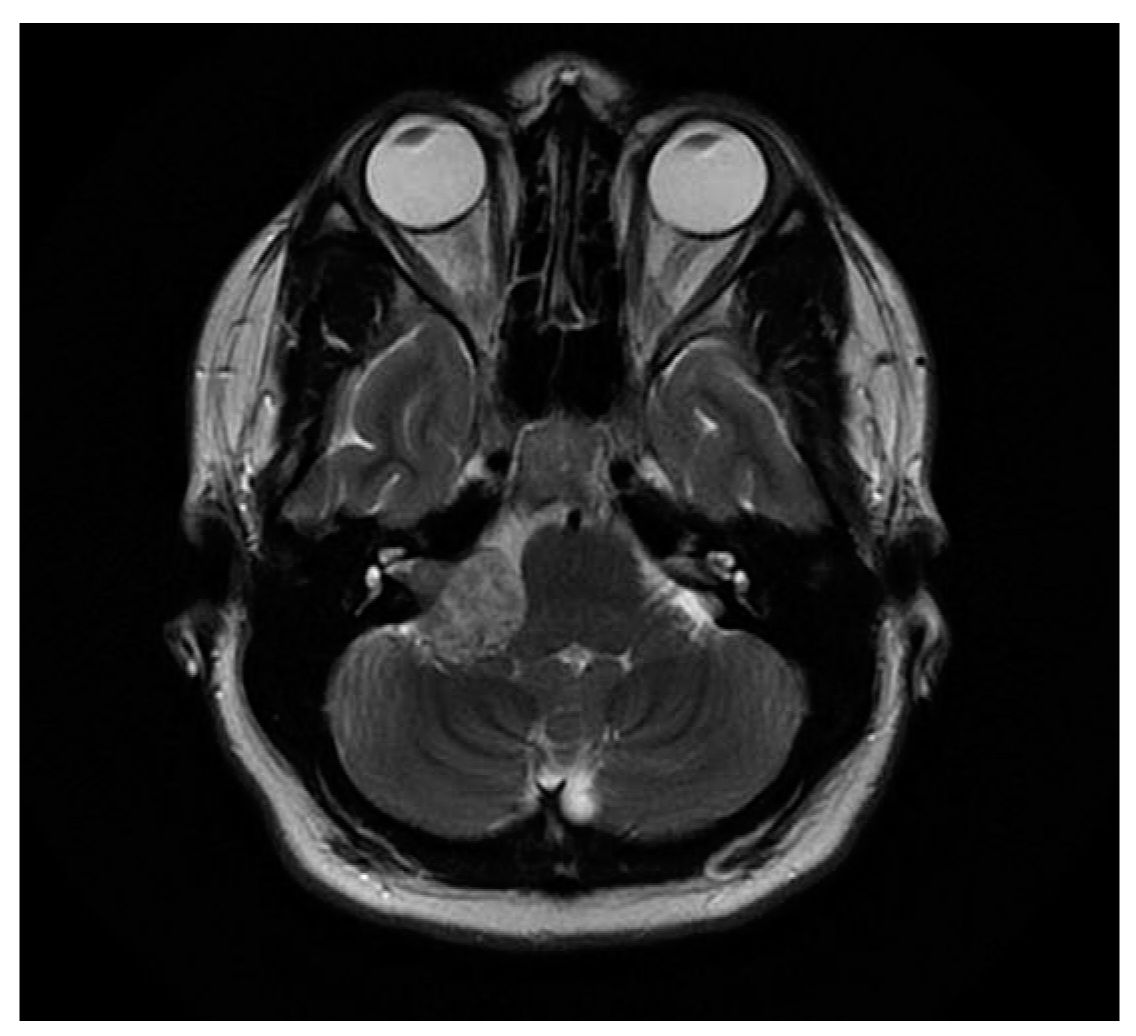
FIGURE 37.3 Brain MRI showing a right vestibular schwannoma in a child with NF2.
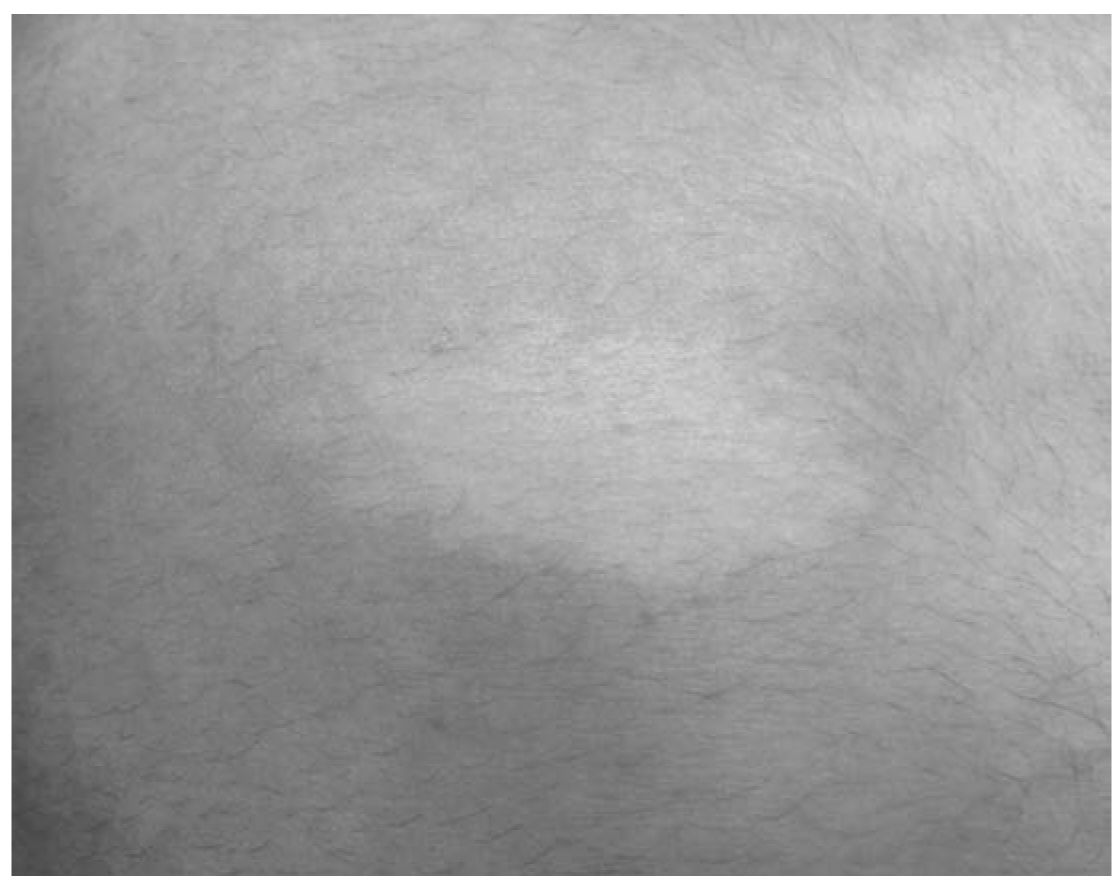
FIGURE 37.4 Hypopigmented macule (ash leaf spot) in a child with TSC. (See color plates.)
I. Evaluation. Children referred for specialty evaluation of developmental delay should have already been screened by their pediatricians. The American Academy of Pediatrics (AAP) recommends such screening utilizing a standardized test at the 9-month, 18-month, and 24- or 30-month well-child care visits. In the absence of such screening, the neurologist should consider administering a standardized general developmental screening test as a preliminary assessment. Examples of validated developmental screening tests include the Ages and Stages Questionnaires, Child Development Inventory, and the Bayley Infant Neurodevelopmental Screen. When screening for autism, consider more specific inventories such as the Childhood Autism Rating Scale (CARS) and the modified checklist for autism in toddlers (M-CHAT). For higher functioning older children, the Autism Spectrum Screening Questionnaire (ASSQ) may be more appropriate.
FIGURE 37.5 Shagreen patch in a child with TSC. (See color plates.)
A complete examination should be done with particular attention to the nervous system. It is especially important to observe and record somatic growth, head circumference, dysmorphic features, birthmarks, and developmental milestones. Audiologic assessment should routinely be obtained in cases of speech and language delays. Referral to a pediatric ophthalmologist is particularly relevant if lack of eye contact, impaired visual tracking, strabismus, and corneal opacities are noted. Most states now routinely screen newborn infants for congenital hypothyroidism and numerous metabolic disorders including phenylketonuria, galactosemia, biotinidase deficiency, and amino acid/urea cycle disorders. However, these should be repeated if warranted by the patient’s presenting signs and symptoms. Screening for lead exposure should be considered if environmental risk factors are identified.
If there is a positive family history of developmental delays or if autism is suspected, genetic testing is indicated. Autistic children should be routinely screened for fragile X syndrome. In addition, chromosome microarray analysis should be obtained with particular attention to duplication at the 15q11–q13 and deletion of 22q11.2 regions. If Rett’s syndrome is a consideration, screening for MeCP2 is recommended.
EEG and MRI are not routinely indicated in cases of static encephalopathies. However, an EEG should be obtained in patients with suspicion of seizures. An EEG should also be considered in patients with regression of language skills to rule out epileptogenic encephalopathies such as Landau–Kleffner syndrome.
Computed tomography (CT) and MRI require sedation and are reserved for patients with abnormalities of head circumference, particularly progressive macrocephaly or microcephaly. Focal or lateralizing neurologic signs (e.g., hemiparesis) are also an indication for imaging studies. Progressive encephalopathies also warrant an MRI to assess for degenerative or structural abnormalities. Periodic neuroimaging is indicated to monitor the progress of children with neurocutaneous syndromes.
Normal motor development follows a defined sequence of milestones. By 2 months of age, the infant lifts the head from prone. Rolling over and transferring objects is accomplished by 5 to 6 months. Sitting independently occurs by 8 to 9 months and walking independently between 12 and 15 months. Delays in the acquisition of milestones are seen in children with hypotonia and weakness. This condition is also described as floppy infant syndrome.
Neuroimaging is indicated if cerebral dysgenesis is suspected. Workup for lower-motor unit disorders includes measurement of creatine kinase level, electromyography/NCV, and muscle biopsy. The edrophonium chloride test can confirm myasthenia gravis (MG). There are now specific DNA probes available for the spinal muscular atrophies, myotonic dystrophy congenital muscular dystrophy, and Prader–Willi syndrome.
A. Central or cerebral hypotonia. Hypotonia may be the initial manifestation of CP. These infants may have a history of hypoxic–ischemic encephalopathy, intraventricular hemorrhage, and/or neonatal seizures. Cerebral dysgenesis should be suspected if there are dysmorphic features or other congenital malformations. Although these infants may initially be hypotonic, over time muscle tone increases accompanied by spasticity, weakness, hyperreflexia, and positive Babinski signs.
Children with chromosome abnormalities and genetic syndromes are also typically hypotonic. Examples include Down’s syndrome, Lowe’s syndrome (oculocerebrorenal syndrome), familial dysautonomia (Riley–Day syndrome), and Prader–Willi syndrome. Prader–Willi syndrome is further characterized by poor feeding in the neonatal period and hypogonadism.
Benign congenital hypotonia may be a mild variant of cerebral hypotonia. Infants with this condition are hypotonic in infancy but gradually recover muscle tone and motor milestones. However, mild developmental delays and learning problems may later be noted in such children.
B. Spinal cord injuries. Stretching of the spinal cord may result from traction during delivery, particularly with breech presentation. The infant is often comatose and flaccid at birth and may not survive. Milder cervical cord injuries present with residual hypotonia and must be distinguished from cerebral and lower-motor unit disorders.
C. Lower-motor unit disorders.
4. Anterior horn cell disorders. Spinal muscular atrophy (SMA) is caused by degeneration of anterior horn cells in the spinal cord and brainstem. SMA type 1 (Werdnig–Hoffman disease) results in severe hypotonia, weakness, absent MSRs, and tongue fasciculations. Affected children usually die from respiratory complications by 1 year of age. Milder variants (SMA types 2 and 3) present later in infancy or childhood; affected children survive longer, but also have weakness, hypotonia, areflexia, muscle fasciculations, and arthrogryposis.
5. Neuropathies. Hereditary neuropathies generally present beyond infancy. They are divided into hereditary motor sensory neuropathy (HMSN), hereditary motor, and hereditary sensory and autonomic subtypes. Charcot–Marie–Tooth, the most common HMSN, presents in childhood with peroneal muscle atrophy foot-drop, pes cavus, and absent MSRs. Neuropathy may also be a manifestation of systemic diseases such as diabetes and autoimmune disorders as well as leukodystrophies and hereditary ataxia. A number of medications including vincristine, isoniazid, phenytoin, and pyridoxine can cause neuropathy (see Chapter 51).
6. Neuromuscular junction disorders. Transient neonatal myasthenia syndrome may be seen in infants born to mothers with MG. Feeding problems, weak cry, facial weakness, and hypotonia are common features. Genetic myasthenic syndromes are rarer and are characterized by respiratory difficulties, poor feeding, weakness, ptosis, and ophthalmoplegia. Infantile botulism mimics MG, but affected infants also show pupillary dilatation and constipation (see Chapter 53).
7. Myopathy. Congenital myopathies present in infancy with hypotonia, motor delays, and proximal muscle weakness. Numerous variants have been described, each with unique findings on muscle biopsy. These include nemaline myopathy, myotubular myopathy, congenital fiber-type disproportion, and central core disease. Metabolic myopathies include lysosomal enzyme deficiencies such as Pompe’s disease (acid maltase deficiency). This disorder presents in infancy with weakness, hypotonia, and cardiomyopathy resulting in heart failure. Rare mitochondrial myopathies can also be accompanied by hypotonia and lactic acidosis. Congenital muscular dystrophy presents with hypotonia and arthrogryposis at birth and may also show CNS involvement. Congenital myotonic dystrophy occurs in infants of mothers with the disease. Hypotonia, facial diplegia, arthrogryphosis, and gastroparesis are the common presenting signs. Childhood-onset muscular dystrophy is reviewed in Chapter 52.
D. Evaluation. Neurologic and general examination helps to distinguish central hypotonia from lower-motor unit disorders. MSRs are present and may be brisk in central hypotonia but are absent or diminished in lower-motor unit disorders. Tongue fasciculations are seen in SMA. Congenital malformations, dysmorphic features, and abnormalities of head size and shape suggest cerebral dysgenesis. Arthrogryposis suggests an SMA or CMD variant.
ATAXIA
A. Definition. Ataxia refers to lack of coordination and impaired control of voluntary movements and balance. The cerebellum controls these functions in conjunction with the sensory input from the vestibular system, basal ganglia, and spinal cord (see Chapter 29).
B. Acute ataxia. Intoxication, particularly from alcohol, sedatives, antihistamines, and anticonvulsants, may cause ataxia. Acute postviral cerebellar ataxia usually occurs in preschool children. The onset often coincides with the end of a viral illness, particularly varicella. The onset is very dramatic with the child suddenly becoming unable to walk. Nystagmus may also occur. Recovery is usually complete but may take up to several months. Viral encephalitis with cerebellar and/or brainstem involvement can also present with acute ataxia. There may be accompanying cranial nerve deficits. GBS (Fisher variant) may present with similar findings. Kinsbourne’s syndrome, characterized by acute ataxia accompanied by opsoclonus and myoclonus, is thought to be a paraneoplastic condition secondary to occult neuroblastoma.
C. Chronic progressive ataxia. Posterior fossa tumors are the most common brain tumors in childhood. Medulloblastoma, ependymoma, cerebellar astrocytoma, and brainstem glioma can all present with slowly progressive ataxia (see Chapter 57). The most common recessive genetic disorder presenting with childhood-onset progressive ataxia is Friedreich ataxia. In addition to ataxia, patients develop scoliosis, cardiomyopathy, retinitis pigmentosa, cataracts, and hearing loss. Absent MSRs and Babinski signs are typically noted. Friedreich ataxia has been localized to an unstable GAA triplet repeat of the frataxin gene on chromosome 9q13. Other rare degenerative and metabolic causes of chronic progressive ataxia include ataxia telangiectasia, abetalipoproteinemia, Refsum’s disease, vitamin E deficiency, biotinidase deficiency, and a multiple carboxylase deficiency. In addition, more than 20 variants of autosomal-dominant spinocerebellar atrophy have been described with ages of onset ranging from childhood to late adulthood.
D. Chronic nonprogressive ataxia. This is typically associated with malformations of the cerebellum and posterior fossa such as Dandy–Walker syndrome, Chiari malformation, and Joubert’s syndrome.
E. Intermittent ataxia. This may accompany concussion and may also be seen as an ictal or postictal feature of epilepsy. Basilar migraine often presents with ataxia and vertigo followed by occipital headache. Benign paroxysmal vertigo presents in early childhood with sudden brief episodes of pallor, nystagmus, and inability to walk. Ataxia presenting acutely or intermittently may also be a feature of childhood-onset multiple sclerosis.
F. Rare genetic causes of episodic ataxia include urea cycle disorders, intermittent maple syrup urine disease, and Hartnup disease. Episodic ataxia type 1 is caused by a dominantly inherited mutation of the voltage-gated K+ channel gene (KCNA1) located at chromosome 12p13. Affected patients present with brief episodes of ataxia sometimes provoked by startle. Myokymia and large calves are associated features. Episodic ataxia type 2 has been linked to a calcium-channel gene (CACNA1A) at chromosome 19p13. Attacks of ataxia begin in childhood. Some are prolonged and resemble basilar migraine.
G. Evaluation. Because ataxia may be congenital, acute, chronic progressive, chronic nonprogressive, or intermittent, it is important to establish the duration and pattern. Inquiry should be made regarding possible antecedent trauma, toxin exposure, medications, infections, and seizure disorder. Developmental delays and/or regression may also accompany ataxia because of many of these disorders.
H. Examination. Truncal ataxia is assessed by checking gait, station, tandem gait, and Romberg sign. Limb ataxia is tested by evaluating finger-to-nose, heel-to-shin, and rapid alternating movements. Ability to perform tests of coordination is age-dependent and also requires the cognitive understanding of the child. In preverbal children, assessment of gait and coordination is done by observation of the child’s movements and activities.
I. Diagnostic studies. Neuroimaging (CT or MRI) is required for most acute ataxias unless intoxication or acute postviral ataxia is determined to be the cause. For acute ataxias, obtain toxicology screening, particularly for alcohol and anticonvulsant drugs, and urine catecholamines. Chronic ataxias also require neuroimaging as well as selective laboratory studies such as urine amino acids, ammonia, phytanic acid, biotin, cholesterol, and cholestanol levels. For the intermittent ataxias, consider selective testing as appropriate for metabolic and genetic disorders (see Chapters 29 and 49).
HEADACHE
A. Acute headache. Headache is a common feature of systemic febrile illnesses and viral syndromes in children. Bacterial illnesses such as otitis media, sinusitis, and meningitis are also accompanied by headaches. Sinusitis may result in facial, frontal, or retro-orbital pain. The pain of otitis media may localize to the temporal region. Meningitis may manifest with diffuse headache, fever, nuchal rigidity, altered mental status, and seizures.
B. The child who presents with acute headache and is afebrile may still have an infectious illness. However, it is more likely that the etiology is the first tension or migraine headache. Head injuries with subarachnoid, subdural, or epidural hemorrhage must be ruled out. Concussions are also typically followed by acute and sometimes chronic headaches. Idiopathic intracranial hypertension (IIH), arterial hypertension, and medication side effects are also considerations.
C. Chronic headache.
1. Migraine is common in childhood affecting up to 5% of preadolescent children and 10% of teenagers. The gender ratio is even in children but females predominate in puberty and adolescence. Family history in a first-degree relative is very common and helps to make the diagnosis. Migraine may appear in early childhood. In such cases, migraine equivalents such as cyclic vomiting, benign paroxysmal torticollis, and benign paroxysmal vertigo are the more likely manifestations. Ophthalmoplegic migraine has also been reported in infancy.
In children, migraines without aura are much more frequent than migraines with aura. The child complains of a moderate to severe diffuse or bifrontal headache accompanied by nausea, vomiting, pallor, and irritability. Older children may be able to better localize the headaches and will describe a throbbing or pulsating quality. The headache may occur at random times and may on occasion awaken the child from sleep. Migraines in children are typically relieved within a few hours by sleep in a darkened room and minor analgesics. Caffeine withdrawal, nitrates, chocolate, monosodium glutamate, alcohol, dairy products, and numerous other foods are thought to trigger migraines. Environmental factors such as secondhand smoke, automobile emissions, perfumes, and atmospheric pressure changes have also been implicated.
Complicated migraines are also seen in childhood and may require further investigations including neuroimaging. Ophthalmoplegic migraine presents with headache and irritability, followed by unilateral third-nerve palsy. This is manifested by ptosis, mydriasis, and eversion of the affected eye, which may last hours to days. Basilar migraine presents with posterior headache, nausea, vomiting, ataxia, vertigo, and on occasion, loss of consciousness. Hemiplegic migraine may mimic stroke with unilateral hemisensory, hemiparetic, and aphasic symptoms followed by a severe contralateral headache.
2. Tension headache. These headaches occur in children as well as adults. The headaches are frontal or occipital or may have a “hatband” distribution. They tend to occur in the afternoon or evening and have been associated with stress or anxiety. When these headaches occur on a daily basis, school avoidance should be suspected. Chronic daily headache is typically seen in adolescent girls and is defined by more than 15 headaches per month. This condition is also sometimes described as “chronic migraine.” Sleep deprivation, skipping meals, excessive gum chewing, smoking, and caffeine withdrawal may exacerbate the headaches. Overuse of analgesics can complicate the situation and result in so-called “rebound” headaches. Biofeedback and/or psychological counseling may be required to treat underlying comorbid conditions such as anxiety, depression, ADHD, and conduct disorders.
Chronic daily headaches caused by mass lesions are associated with increasing intracranial pressure. Abnormalities on neurologic examination appear in the great majority of such children within 4 months of the onset of headaches. These may include papilledema, cranial nerve abnormalities, ataxia, dysmetria, hemiparesis, or focal sensory signs. Reflex asymmetries and a unilateral Babinski sign may also be present.
IIH presents with acute or chronic headaches, vomiting, and double vision accompanied by papilledema. It is commonly seen in obese adolescent girls. A sixth nerve paresis may also be noted. Lumbar puncture (LP) demonstrates elevated cerebrospinal fluid opening pressure and may partially relieve the condition. The mechanism of action is unknown, but IIH has been associated with use of corticosteroids, vitamin A, and tetracyclines. Cerebral venous thrombosis and mastoiditis have also been linked to the condition.
D. Evaluation. It is important to assess the frequency, severity, location, and time of day of the headaches. Severity can be assessed by asking the child or parent to grade the headache from 1 to 10. The history should include inquiry regarding development, head injuries, seizures, learning or attention problems, and family members with recurrent headaches. Information should be requested regarding lifestyle factors including caffeine consumption, sleep habits, meal patterns, excessive gum chewing, and exposure to secondhand smoke. In girls, the onset of menarche should be noted because it may be heralded by migraine headaches.
Blood pressure and pulse should be checked personally by the physician or a reliable assistant. A complete general and neurologic examination is required for every child who presents with headaches. The head circumference should be measured. The skull and neck should be auscultated for bruits. The eyes, ears, nose, and throat should be examined including palpation for cervical nodes and sinus tenderness. The temporomandibular joint should be palpated and auscultated for misalignments and clicks. The teeth should be checked for caries, malocclusions, and newly installed braces or appliances.
Most children with headaches do not require neuroimaging provided rapport is established with the parents and timely follow-up can be arranged. If the history and examination suggest acute CNS infection or IIH, an LP should be considered. Acute hypertension may require hospitalization and workup for renal or cardiac diseases. If acute head injury or concussion is suspected, an emergency noncontrast CT scan should be obtained to check for intracranial hemorrhage. Chronic recurrent headaches with typical migrainous or tension features do not require imaging unless they remain refractory to treatment. An MRI should be obtained in children with complicated migraine and IIH or if abnormalities are present on neurologic examination. On occasion, an anxious parent may insist on neuroimaging for the child despite the reassurance that such a procedure is unnecessary. In such instances, it is usually prudent to acquiesce to the parent’s request provided there is no obvious contraindication to the neuroimaging procedure. The reader is referred to Chapters 20, 21, and 54 for further information on the approach to patients with acute and chronic headaches.
NONEPILEPTIC PAROXYSMAL DISORDERS
Nonepileptic paroxysmal disorders are defined by their intermittent, recurrent, and abrupt presentation. Between episodes the patient feels well and has no complaints. It is particularly important to distinguish these episodic conditions from epilepsy. Childhood epilepsies are discussed in detail in Chapter 42.
A. Evaluation. A detailed description of the event by a reliable observer is essential for proper classification and diagnosis. Older children can also contribute their personal recollections. Inquiry should be particularly directed toward triggering events or precipitating factors. A videotape of the episode may be particularly useful. Examination is normal in most cases. Diagnostic studies are generally not required. On occasions, EEG and video EEG may be necessary to rule out epilepsy.
B. Breath-holding spells. These are benign, but frightening, paroxysmal episodes that may result in a brief loss of consciousness. Cyanotic syncope occurs in infants and children between 6 months and 4 years of age. The family history is often positive. The child first cries because of anxiety, frustration, or pain. Following prolonged expiration, breathing ceases and the child becomes cyanotic, hypotonic, and briefly unresponsive. Recovery usually occurs quickly but on occasion some brief tonic–clonic movements followed by sleep may occur.
Another variant of breath-holding is pallid syncope. This is usually initiated by a minor head injury, which is thought to trigger a brief reflex asystole. The child becomes pale, limp, and is briefly unconscious.
Breath-holding spells can be diagnosed clinically by the stereotypic sequence of events preceding each episode. On rare occasions or to lessen parental anxiety, an EEG can be done to distinguish these episodes from epileptic seizures.
C. Syncope (fainting). Syncope is common in children, especially during adolescence. It is induced by a transient decrease in blood flow to the brain secondary to a vasovagal reflex. It is almost always precipitated by triggering stimuli such as change of position, pain, or extreme fear or anxiety.
The syncopal event may be preceded by an “aura” of blurry vision, dizziness, and/or tinnitus. The child then becomes pale and clammy and loses consciousness with an accompanying fall. Brief stiffening, upward eye rolling, vocalizations, and tremulous movements are not uncommon. Tongue biting is unusual in syncope. Recovery of consciousness usually occurs within a minute. In most instances, syncope is considered to be a common benign occurrence that does not warrant extensive neurodiagnostic testing. If syncope is prolonged or recurrent, a cardiology referral with ECG monitoring may be necessary. Syncope is reviewed in more detail in Chapter 7.
D. Other. Episodic weakness, ataxia, tremor, chorea, vertigo, and sensory disturbances are reviewed elsewhere in this chapter or in other chapters under the appropriate headings. However, several other paroxysmal neurologic disorders are unique to infants and young children.
1. Spasmus nutans begins in infancy and resolves spontaneously in early childhood. It consists of three cardinal features: head bobbing, torticollis, and nystagmus. Diagnosis may warrant neuroimaging of the brain and orbits to rule out neoplasms, which can rarely present with similar signs.
2. Sandifer’s syndrome is seen in infancy and consists of paroxysmal opisthotonic posturing sometimes accompanied by vomiting and apnea. The underlying etiology is gastroesophageal reflux, which requires pediatric gastrointestinal consultation.
3. Paroxysmal infant shuddering or shivering occurs during wakefulness and may raise concerns about seizures. The child appears to momentarily shiver as if having a chill. Such episodes often occur frequently enough to be captured on videotape in which case they are easily distinguishable from clinical seizures. Later in life, essential tremor may be associated with a history of infant shuddering.
4. Gratification syndrome (masturbation) is occasionally seen in infants and young children and may be confused with seizures. These episodes consist of prolonged rocking movements of the pelvis and thighs. Altered breathing, flushing, sweating, and staring are typically seen. These episodes resolve over time with parental reassurance.
A. Normocephaly is defined as a head circumference measurement between the 2nd and 98th percentile. A head circumference greater than the 98th percentile is macrocephaly, and a head circumference less than the 2nd percentile is microcephaly. Normally the head should grow on a specific percentile. Deviation of growth from the expected percentile should prompt further evaluation and investigations. Macrocephaly and microcephaly are both associated with higher incidences of CNS pathology and developmental delays.
B. Macrocephaly may be familial, in which case at least one parent’s head will also be large. Differential diagnosis also includes hydrocephalus, which may be communicating or obstructive. Rare metabolic disorders including galactosemia, maple syrup urine disease, mucopolysaccharidoses, and Canavan’s disease are associated with macrocephaly. Children with genetic conditions including achondroplasia, fragile X syndrome, cerebral gigantism (Sotos syndrome), and the neurocutaneous disorders commonly have large heads. Benign subdural collections present with progressive enlargement of head circumference, but without signs of increased intracranial pressure.
C. Microcephaly may be secondary to genetic causes or cerebral dysgenesis and is commonly associated with congenital syndromes. It is also frequently seen following hypoxic–ischemic encephalopathy, intrauterine infections, and postnatal infection. Craniosynostosis can cause progressive microcephaly when more than one suture is affected. Rett’s syndrome also presents with progressive microcephaly.
D. Evaluation. The history may reveal developmental delay or regression. Symptoms of increased intracranial pressure include emesis, irritability, and somnolence. Examination may demonstrate signs of increased intracranial pressure including bulging fontanelle, sunsetting of the eyes, papilledema, or a sixth nerve palsy. The head circumference should be plotted on a standardized growth chart and serial measurements should be obtained. Neuroimaging is indicated in most cases of abnormal head size. Exceptions include familial macrocephaly and possibly benign subdural collections. Genetic and metabolic studies are also considerations if cerebral dysgenesis or storage diseases are suspected.
POSTCONCUSSION SYNDROME
Concussions are biomechanical brain injuries that occur following low-velocity impacts to the head, face, and neck. They are common in children and adolescents who participate in contact sports. In the United States about 300,000 athletic concussions occur each year. Concussions are most commonly seen in football, but also in girls’ soccer and basketball and boys’ wrestling and hockey. All 50 states have passed legislation mandating education, prevention, and medical supervision of sports-related concussions (SRC). The American Academy of Neurology (AAN), AAP, and American Medical Association (AMA) have all issued position statements regarding management of SRC and return-to-play (RTP) guidelines.
Symptoms of postconcussion syndrome include headache, memory problems, nausea, vomiting, altered sensorium, emotional lability, impaired concentration, and sleep disturbances. Fewer than 10% of SRC are associated with an immediate loss of consciousness. If a concussion is suspected, the athlete should be immediately removed from play and evaluated on the side line by medically trained personnel utilizing a standardized symptom checklist. Loss of consciousness, retrograde amnesia, a post-traumatic seizure, or a Glasgow Coma Scale score <15 warrants immediate physician evaluation.
Neurologic consultation is appropriate in determining the initial severity a concussion and whether neuroimaging is required. Subsequent management of postconcussion syndrome consists mainly of closely monitored physical and cognitive rest until all symptoms have resolved. Extra caution is required if the patient has a history of multiple concussions or if symptoms persist for longer than 10 days. Neuropsychological testing may be indicated in these situations. Alternatively, serial computerized neurocognitive testing programs may be utilized. RTP should be stepwise and carefully supervised by the physician in cooperation with coaches and trainers. Persistent headaches, vertigo, or concentration problems may require long-term neurologic follow-up.
• ADHD has an incidence of 11% in childhood. ADHD, predominantly hyperactive-impulsive, ADHD, predominantly inattentive, and ADHD combined are the three main types.
• Developmental delay affects 5% to 10% of children and is frequently caused by static encephalopathies including CP and ASDs.
• The definition of ASDs has been dramatically revised by DSM5. Social-pragmatic communication disorder is a new diagnostic category and pervasive developmental disorder and Asperger’s syndrome have been eliminated from DSM5.
• Neurocutaneous syndromes are neurologic disorders with skin lesions and CNS findings. NF1 and TSC are two types that can present with CNS tumors.
• Headaches are not uncommon in children. Pediatric migraines typically present without aura. There are also migraine variants unique to children including cyclic vomiting and benign paroxysmal torticollis.
• Nonepileptic paroxysmal disorders are characterized by their intermittent recurrent and abrupt presentation. Examples include breath-holding spells, spasms nutans, and Sandifer’s syndrome.
• In the United States, about 300,000 athletic concussions occur annually. Guidelines for management and RTP have been issued by the AAN, AAP, and AMA.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


