FIGURE 32.1 Baroreflex pathways. Efferents from arterial carotid sinus baroreceptors travel to the CNS with the glossopharyngeal (IX) nerve and those from the arterial aortic arch baroreceptors travel with the vagus (X) nerve. S preganglionic efferents travel in the intermediolateral cell column of the spinal cord (IML) to synapse with postganglionic neurons (nicotinic acetylcholine receptors) of the S ganglia. Postganglionic neurons travel to innervate vascular smooth muscle, the heart, and the adrenal glands. Vagal preganglionic neurons are situated in the ventrolateral medulla and the dorsal vagal nucleus and vagal innervation of the cardiac sinus node travels with the vagus nerve. The baroreflex. 1, Standing produces a shift or “pooling” of 600–1000 mL of blood to the lower body (mainly limb and splanchnic capacitance circulation), reducing venous return and cardiac output; 2, This is sensed by specialized stretch receptors (arterial and cardiopulmonary baroreceptors) that in turn activate (unload) baroreflexes. Inputs from carotid and aortic baroreceptors travel with the glossopharyngeal (IX) and vagus nerves (X) to converge on cardiovascular centers in the brainstem and medulla (mainly nucleus of the tractus solitarius) and their projections; 3, The physiologic baroreceptor reflex response to the volume shifts produced by upright posture is a compensatory increase in S tone with a decrease in PS outflow; 4, S nerve terminals release NE that produces increased vasoconstriction of skeletal and mesenteric muscle vessels, HR, and cardiac contractility. Additional increases in venous return occur through a “pumping” effect of contracting limb and abdominal muscles engaged by the effort of standing (not shown); 5, A longer-term response promoting extracellular fluid volume expansion includes baroreflex-mediated release of renin, angiotensin, and aldosterone, leading to increased renal Na++ absorption, and release of vasopressin, with an increase in free-water absorption. A, Afferent baroreflex limb dysfunction is present when lesions involve baroreceptors and IX and X cranial nerves (neck surgery, radiotherapy, trauma, neuropathies, and autonomic disorders such as baroreflex failure, Holmes–Adie syndrome, and HSAN III); B, Central lesions involve the ventrolateral medulla (MSA), descending S pathways (medullary lesions, spinal cord lesions above T5), and IML (MSA, Lewy body disorders); C. S ganglia lesions involve S ganglia (autoimmune ganglionopathies associated with nAChR antibodies or paraneoplastic, Lewy body disorders); D, E, Postganglionic S and efferent PS lesions are present with small-fiber neuropathies (diabetes, amyloidosis, Sjögren’s syndrome, HSAN III). F: Efferent S neuroeffector junction dysfunction occurs with dopamine β-hydroxylase deficiency and α-1 adrenoceptor blocking drugs. AVP, arginine vasopressin; CNS, central nervous system; CVM, caudal ventrolateral medulla; HR, heart rate; IML, intermediolateral cell columns of the spinal cord; NA, nucleus ambiguus; MSA, multiple system atrophy; nAChR, nicotinic acetylcholine receptor antibodies; NE, norepinephrine; NTS, nucleus tractus solitarius; RVM, rostral ventrolateral medulla; S, sympathetic; SA, sinus node.

C. Autonomic testing. ANS testing is an extension of the physical examination. Conclusions arising from ANS testing are of most value when the selection of tests is guided by the clinical findings. As an abnormal test does not always imply disease, several tests are usually required to confirm autonomic dysfunction. Combing several autonomic test results and BP values into a composite scale (composite autonomic scoring scale) has been used as a reliable measure of autonomic function. Autonomic labs should have standardized testing conditions and normative test data.
Most ANS tests assess the integrity of an autonomic reflex indirectly by recording stimulus-evoked effector organ responses. Bedside ANS tests (Table 32.3) measure those effector organ responses that are most easily recorded (i.e., changes in HR, BP, pupillary size, sweating patterns, SSR, and so on). Sophisticated tests such beat-to-beat arterial pressure recordings during different challenges (i.e., changes in posture, Valsalva-like and other maneuvers— ![]() Video 32.1) and measurement of sweat output and its latencies (i.e., quantitative sudomotor axon reflex test [QSART]), to assess efferent sudomotor function, require more complex and expensive instruments.
Video 32.1) and measurement of sweat output and its latencies (i.e., quantitative sudomotor axon reflex test [QSART]), to assess efferent sudomotor function, require more complex and expensive instruments.
The release of autonomic neurotransmitters (and their metabolic byproducts) in response to different challenges (i.e., by maneuvers, changes in posture, pharmacologic stimuli) may be helpful in some diagnostic situations (Table 32.4).
Cardiac S innervation, as measured by the uptake of radionuclide tracers (i.e., 123-meta-iodo-benzylguanidine—a guanethidine analog) into cardiac S neurons, helps discriminate central (it is preserved with MSA) from peripheral ANS dysfunction (it is impaired with ANS peripheral neuropathies, pure autonomic failure (PAF), Lewy body dementia (LBD), Parkinson’s disease (PD) patients with OH, and in some PD patients without OH).
D. Laboratory and other evaluations.
1. Screening tests.
a. Complete blood count
b. Erythrocyte sedimentation rate
c. Blood urea nitrogen (BUN)/creatinine (Cr)
d. Glucose, fasting (hemoglobin A1C, glucose tolerance test if diabetes suspected)
e. Thyroid-stimulating hormone
f. Vitamin B12/folate
g. Immunoelectrophoresis/immunofixation (serum and urine)
h. Urinalysis (U/A)
i. Human immunodeficiency virus (HIV)
j. Testosterone levels (in patients with erectile dysfunction [ED])
2. Additional testing is usually directed by the clinical presentation.
a. Autoantibodies.
(1) Ganglionic nicotinic a 3 acetylcholine receptor antibodies (a 3-AChR) (see section Autonomic Neuropathies, below), paraneoplastic autoantibodies (anti-Hu [ANNA-1]), and voltage-gated P/Q type and N-type calcium channel and potassium channel [VGKC], in particular with autonomic symptoms of subacute onset. Anti-Hu and anti-CV2/collapsin response mediator protein 5 (CV2/CRMP-5) are frequently present in autonomic neuropathies.
(2) VGKC and anti-N-methyl-d-aspartate antibodies when limbic encephalitis is present.
(3) Antinuclear antibodies, rheumatoid factor, anti-Ro/SS-A, and anti-La/SS-B (Sjögren’s, and other connective tissue disorders) when autonomic dysfunction is associated with somatic peripheral neuropathy.
b. Electrophysiologic studies. Nerve conduction studies (NCS) and electromyography (EMG), sphincter and pelvic floor EMG to detect denervation potentials of spinal cord anterior horn cells (MSA), polysomnography (MSA), and urodynamic studies.
c. Imaging studies include magnetic resonance imaging (MRI) of brain and spine, which determine CNS and spinal cord lesions and pelvic imaging structural lesions.
d. Histopathologic tests include amyloid staining in fat aspirate, rectal or gingival biopsy, intraepidermal sweat gland nerve fiber density, and sural nerve biopsy.
e. Other testing includes aminolevulinic acid, porphobilinogen, and porphyrins (24-hour urine collection), erythrocyte porphobilinogen deaminase activity (porphyria), testing for deficient α-galactosidase A enzyme activity in males, and genetic testing (inherited neuropathies).

SELECTED CLINICAL PRESENTATIONS OF AUTONOMIC DYSFUNCTION
Orthostatic Hypotension and Orthostatic Intolerance Syndromes
NEUROGENIC ORTHOSTATIC (POSTURAL) HYPOTENSION
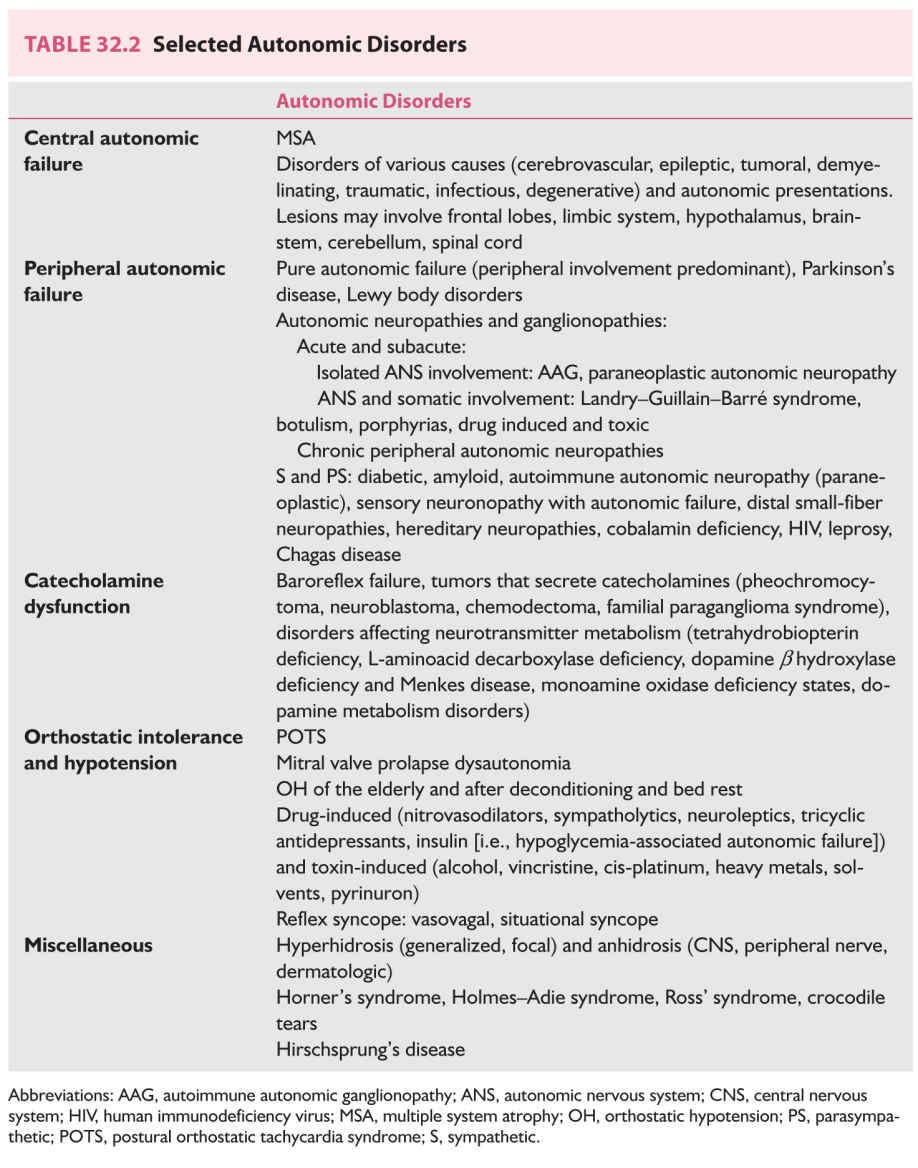
Orthostatic hypotension (OH), defined as a sustained decrease in systolic pressure of at least 20 mm Hg (30 mm Hg in patients with supine hypertension) and of diastolic pressure of at least 10 mm Hg within 3 minutes of standing or head up tilt to at least 60 degrees on a tilt table, is a frequent and disabling manifestation of autonomic disorders. It may be the initial sign (i.e., “tip of the iceberg”), heralding the onset of autonomic disorders.
A. Diagnosis. BP measured with a sphygmomanometer and pulse rate recorded while in supine rest (once BP values are stable) and after standing for 2 to 3 minutes determine if OH is present. HR responses add valuable data and help determine a diagnosis (Table 32.3). OH standing times (the maximum time a patient is able to stand) replace BP in patients with severe OH who do not tolerate standing for enough time to allow BP measurement.
B. Pathophysiology. Hemodynamic responses to shifts in volume produced by changes in posture are complex, and depend on intact baroreflexes (Fig. 32.1). Neurogenic orthostatic hypotension (NOH) is due to deficient vasoconstriction, occurring because of failure to appropriately release norepinephrine (NE), the S postganglionic neurotransmitter that innervates blood vessels. This may be due to impaired afferent baroreflex pathways or to impaired efferent S outflow at central or peripheral sites. (Chronic OH due to autonomic failure should not be confused with episodic hypotension caused by the acute reflex withdrawal of S vascular tone occurring with vasovagal syncope—see section Reflex Syncope, Neurally Mediated Hypotension, and Bradycardia.)
C. Causes.
1. Medications (in particular antihypertensive agents)
2. Autonomic neuropathies
3. Spinal cord lesions, above T4 or T5 (complete transection and higher levels impair more S efferent nerves).
4. Brainstem and medulla lesions
5. MSA, PD, dementia with Lewy bodies (diffuse Lewy body disease [DLB]), and PAF should be considered in the differential diagnosis of OH (Fig. 32.1).
6. Exacerbating factors. Extracellular volume depletion (dehydration), physical deconditioning, and aging.
D. Treatment.
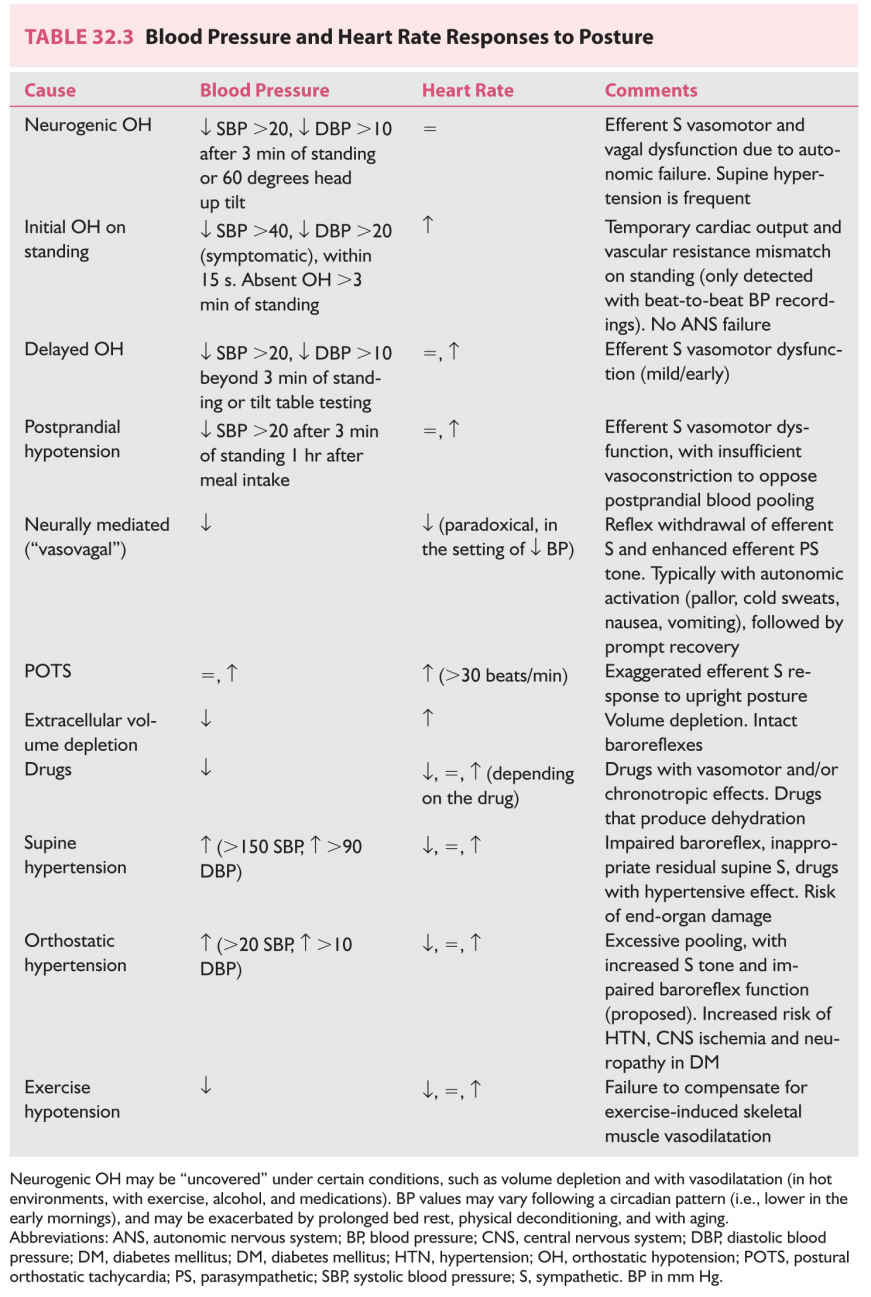
Treatment of OH is directed at improving a patient’s symptoms and functional capacity and quality of life, rather than treating BP “numbers.” Recording symptoms and BP and HR while supine and after standing for >2 to 3 minutes, before and 1 hour after meals, to determine if postprandial (postcibal) hypotension is present, facilitates management. Neurally mediated (vasovagal) hypotension and bradycardia, postural orthostatic tachycardia, OH in the elderly, orthostatic intolerance associated with prolonged bed rest, and deconditioning and spaceflight also respond to these treatments.
7. Nonpharmacologic treatment (Table 32.5, Video 32.1) is indicated for all patients with orthostatic intolerance (i.e., NOH, vasovagal syncope, and postural tachycardia). Countermeasures are used to acutely raise BP when symptoms of cerebral hypoperfusion are present.
8. Pharmacologic treatment (Table 32.6). Fludrocortisone acetate is considered the first-line drug in the treatment of OH.
Drugs with vasoconstrictor effects are administered during the patient’s active hours (and withheld in evenings) to minimize supine hypertension. Supine hypertension is a manifestation of impairment of the baroreflex, with failure to prevent excessive BP rise in response to increased venous return when supine. It is sometimes severe enough to lead to hypertensive end-organ damage. Recommendations include avoiding supine postures, always sleeping with the head of the bed elevated (reverse Trendelenburg ~30 degrees), and having bedtime meals (to produce postprandial BP drop). Short-acting antihypertensive drugs (nitroglycerine, nifedipine, losartan, clonidine) are helpful.
POSTURAL ORTHOSTATIC TACHYCARDIA SYNDROME
Postural orthostatic tachycardia syndrome (POTS) is characterized by excessive tachycardic response to upright postures (Table 32.3). It may be accompanied by various ANS, psychiatric, and somatic complaints. Some forms are associated with alterations in adrenergic (sudomotor and vascular tone) function (pheochromocytoma should be considered). Many hemodynamic features are similar to those of deconditioning produced by bed rest and exposure to microgravity.

REFLEX SYNCOPE, NEURALLY MEDIATED HYPOTENSION, AND BRADYCARDIA
Reflex syncope is a short-duration, self-terminating loss of consciousness that results from a chain of ANS events (i.e., neurally mediated) that lead to hypotension and cerebral hypoperfusion. Onset is rapid and is followed by spontaneous, prompt, and complete recovery. Vasovagal syncope is the most common cause of reflex syncope. Prodromal symptoms of autonomic activation (sweating, pallor, nausea) are typical. Vasovagal syncope is reflexly triggered by emotion (i.e., emotional syncope) and by gravitational volume shifts produced by upright postures. Other causes of reflex syncope are situational (i.e., cough, micturition). All forms of reflex syncope require relatively preserved PS and S pathways as the typical hemodynamic response involves reflex vasodilatation (due to S withdrawal) and slowing of the HR (due to PS activation via the vagus nerve). The diagnosis is clinical and cardiac (and sometimes epileptic) causes should be considered. Treatment includes educating the patient to avoid triggering situations and how to apply some of the counter-maneuvers outlined above for OH. Some medications may be effective in reducing the risk of recurrent reflex syncope.
SEXUAL DYSFUNCTION AND URINARY BLADDER DYSFUNCTION
A wide range of neurologic diseases are associated with sexual and urinary bladder dysfunction.
SEXUAL DYSFUNCTION
Male and female sexual dysfunction is common. Sexual dysfunction may manifest as disorders of libido, of arousal and orgasm in women, and of erection and ejaculation in men. Sexual dysfunction in men may be an early sign of autonomic dysfunction, first with ED (discussed here) and later by failure of ejaculation.
Pathophysiology
The sexual response cycle of excitement, plateau, orgasm, and resolution is mediated through the integrated and coordinated activity of the somatic and ANSs. Central ANS networks and pathways involved in controlling sexual function include the medial preoptic area, amygdala, periventricular nucleus, periaqueductal gray, and the ventral tegmentum. They project onto S fibers originating from T12–L2 that synapse in the hypogastric plexus to travel to the pelvic plexus, where they meet PS fibers from S2–S4 segments. From here, S and PS travel together in pelvic nerves to innervate the pelvic organs.
Sexual response cycles begin with smooth muscle relaxation with vasodilatation and an increase in pelvic blood flow and genital engorgement. Genital engorgement is a neurovascular event dependent on spinal autonomic centers, enhanced by genital stimulation and by supraspinal sexual centers. Ejaculation depends on two different spinal reflexes. One results in emission of semen into the urethra, and another, in ejaculation.
Erectile Dysfunction
A. Diagnosis. ED is the persistent inability to develop and maintain an erection sufficient enough to permit satisfactory sexual performance. ED is a common and sometimes early presentation of autonomic dysfunction. In addition to autonomic causes, ED may result as a side effect of medications, or because of mechanical, endothelial, endocrine, metabolic, vascular, and psychogenic causes, frequently acting in combination. Depression and anxiety are common causes of sexual dysfunction and frequent complicating factors. Particular attention should be paid to the history of symptoms of leg claudication and cardiovascular symptoms as ED is a strong predictor of future cardiovascular disease in younger men. Abdominal examination is necessary as 1% of patients with ED have abdominal aortic aneurysm.
B. Physiology. Penile engorgement and erection result from endothelium-mediated engorgement of the corpora cavernosa (and less so, of the corpora spongiosum) with venous blood. Erection is triggered by somatosensory stimulation (reflexive erection, disrupted by lesions of the sacral cord and intermediolateral cell columns—as in MSA), audiovisual stimulation (psychogenic erection, disrupted by lesions above the sacral cord), and by nocturnal cortical stimulation during rapid eye movement (REM) sleep (“morning erection,” nocturnal penile tumescence, disrupted by disturbed REM sleep).
For erection to occur, appropriate stimuli trigger the release of neurotransmitters from efferent cholinergic PS and noncholinergic, nonadrenergic fibers originating from the pelvic plexus. Key to erection is the release of nitric oxide (NO) from nitrergic nerves in the corpora cavernosa and from cavernous vascular endothelium. NO results in an increased cyclic guanosine monophosphate (cGMP)-dependent dilatation of trabecular arteries. Dilated trabecular arteries in turn compress the cavernous vein against the tunica albuginea, obstructing venous outflow to produce engorgement of the penis with venous blood. Concurrent contractions of the ischiocavernosus and bulbospongiosus muscles compress corpora cavernosa veins to limit venous outflow and maintain erection. Most ED results from deficient release of NO. Penile detumescence occurs when corpus cavernosa phosphodiesterase-5 (PDE-5) hydrolyzes cGMP, ending arteriolar vasodilatation. Understanding of this pathway has led to treatment of ED by using reversible competitive inhibitors of PDE-5 (see section D.2 under Erectile Dysfunction, Sexual Dysfunction below).
C. Laboratory testing. In men who do not respond to treatment with PDE-5 inhibitors, when specific neurologic causes are considered, or when the cause of ED is not apparent:
1. Free testosterone, luteinizing and follicle-stimulating hormone and prolactin levels, glucose tolerance, liver function, prostatic specific antigen, BUN, creatinine, and thyroid function tests.
2. Consultations. Psychiatric, urologic, and vascular consultations in the appropriate clinical settings.
D. Treatment of ED.
1. Nonpharmacologic treatment.
a. Lifestyle changes. Exercise (>18 MET/hour/week), weight loss (goal: BMI <30 kg/m2), and smoking and alcohol cessation are recommended for all patients with sexual dysfunction. Pelvic floor muscle strengthening exercises may be helpful.
b. b. Treatment of underlying endocrine, metabolic, vascular, and psychogenic causes, and management or withdrawal of any offending medications. Psychosexual counseling is important in many cases.
2. Pharmacologic treatment. PDE-5 (see section B under Erectile Dysfunction above) inhibitors are the mainstay of the pharmacologic treatment of ED. By blocking the destruction of cGMP, they potentiate penile vessel smooth muscle relaxation and, thus, penile engorgement. PDE-5 inhibitors require sexual stimulation to be effective (they do not increase libido). PDE-5 inhibitors are not effective where ED is due to significant endothelial dysfunction with deficient NO bioavailability. NO-mediated smooth muscle relaxation is androgen-dependent and supplementing testosterone in those who are deficient (i.e., morning testosterone <300 ng/dL) may offer benefit.
Sildenafil (50 to 100 mg) and vardenafil (10 to 20 mg) taken 1 hour prior to intercourse have a duration of effect up of to 4 hours. Tadalafil (10 to 20 mg), taken 1 to 12 hours prior to intercourse, has a duration of effect of up to 36 hours. As it has a long half-life, tadalafil should be avoided in patients in whom cardiac risk from sexual activity is high. As interactions between PDE-5 inhibitors and NO donors (i.e., nitrates) may precipitate serious hypotension, the ongoing use of nitrates is an absolute contraindication to their use. Significant hypotension may result from the use of PDE-5 inhibitors in patients with MSA.
3. Local treatments. Used in patients in whom PDE-5 inhibitors are ineffective or contraindicated. Vacuum constriction devices and constriction rings applied to the base of the penis produce unnatural but effective erections. Alprostadil, a prostaglandin E-1 (PGE-1) analog inserted into the urethra, produces penile smooth muscle relaxation and penile erection. Medications (papaverine, alprostadil, phentolamine, and PGE-1) self-injected into the corpus cavernosa to cause an erection are very effective. A normal erection in response to the intracavernosal injection of vasoactive agents (i.e., papaverine) indicates that vascular mechanisms involved in erection are intact, suggesting psychogenic impotence (although approximately 30% of men with normal erections may lack a response). Penile implants are beneficial and well tolerated and are usually reserved for those who PDE-5 inhibitors and other methods are unsatisfactory.
URINARY BLADDER DYSFUNCTION
A wide range of neurologic diseases are associated with urinary bladder dysfunction. Common conditions are stroke, dementia, PD, multiple sclerosis, DM, and other forms of neuropathy with autonomic involvement. Tumoral, infectious, traumatic, and congenital (i.e., spina bifida, tethered cord) causes are less frequent.
Anatomy
The cerebral cortex, basal ganglia, cerebellum, and brainstem pontine detrusor nuclei influence the sacral spinal nuclei involved in urinary bladder control via peripheral nerves. Peripheral nerves innervate the external sphincter, periurethral muscles, and other abdominal and pelvic muscles. Micturition relies on a brainstem and spinal cord autonomic reflex that includes periaqueductal gray and the pontine micturition center (PMC), regulated by the hypothalamus and prefrontal cortex and their inputs from other areas (i.e., with the right insula providing interoception and limbic system providing hedonic valance). Micturition is initiated by the hypothalamus and prefrontal cortex (i.e., consciousness about the appropriateness of voiding) by withdrawing their inhibition of the PMC. The PMC activates relaxation of the external sphincter striated muscle followed by relaxation of the bladder detrusor muscle. Urinary storage depends on a sacral cord autonomic reflex arc. It is tonically facilitated by the pontine storage center, hypothalamus, cerebellum, basal ganglia, and frontal cortex.
Bladder filling triggers the micturition reflex. Micturition is primarily a PS function with S2–S4 efferents stimulating bladder (detrusor muscle) contraction while inhibiting the internal bladder sphincter. The S nervous system is involved in urine storage and bladder capacity. S T10–L2 efferents inhibit the detrusor while stimulating the internal sphincter. Bladder contraction occurs with stimulation of PS cholinergic muscarinic receptors and relaxation with stimulation of b -adrenoceptors. Contraction of the urethra occurs with stimulation of a –adrenoceptors and relaxation with stimulation of nicotinic acetylcholine receptors.
A. History. Patterns of urinary incontinence and frequency and urgency of urination are determined. Desire to void, ability to initiate and terminate urination, force of urinary stream and urine volume, and sensations associated with urination are other important aspects of the history. Particular attention must also be paid to medication.
B. Examination.
The neurologic examination and the postvoid urine residual, complemented by urodynamic studies, determine the site of an autonomic lesion.
1. Lesion sites.
a. Lesions above the PMC produce an uninhibited bladder. There is lack of awareness of bladder filling and of the passage of urine. There is urinary retention with elevated residual bladder urine volumes.
b. Lesions below PMC and above the S2–S4 produce an upper motor neuron bladder. There is lack of awareness of urinary sensations. Bladder and sphincter contractions occur simultaneously (detrusor-sphincter dyssynergia). Residual bladder urine volumes are low, and detrusor is hypertrophic (may produce vesicourethral reflux). With thoracolumbar spinal cord lesions there is loss of S control of the bladder neck resulting in retrograde ejaculation. Spinal cord lesions above T6 result in disinhibition of S neurons and patients with these lesions are at risk of autonomic dysreflexia (see section B under Autonomic Crisis below). With lesions above S2–S4, the voiding reflex is preserved, and there is frequency of micturition, and reduced bladder capacity.
c. Sacral cord or sacral nerve root lesions produce a lower motor neuron bladder. The bladder is flaccid (the PS nucleus is affected), and bladder residual volumes are elevated. There is stress incontinence, and lack of awareness of urinary sensations (although hypogastric nerves transmit pain from a distended bladder).
d. d. Peripheral nerve lesions produce a flaccid bladder (the PS nucleus connections with the bladder are affected), and bladder residual volumes are elevated. There is stress incontinence. If the motor nerves are preferentially involved, volitional voiding may be severely compromised even though bladder sensation may be largely preserved.
C. Evaluation of urinary dysfunction.
1. Urinalysis and urine culture with appropriate sensitivity studies (many patients have associated urinary tract infection), postvoid urine residual. Renal function is monitored as vesicouretheral reflux may cause renal failure.
2. Ultrasound determines postvoid residual and anatomy.
3. Urodynamic investigations. Cystometry provides information about the pressure–volume relation on filling (bladder compliance), bladder capacity, volume at first sensation and at urge to void, voiding pressure, and if uninhibited detrusor contractions are present (Table 32.7). Endoscopy allows direct visualization of the bladder neck during voiding.
4. Spinal and brain imaging, as directed by the clinical features.
5. Sphincter and pelvic floor EMG detects denervation potentials in selected muscles in lesions of the anterior horn cells in the S2–S4 spinal cord, and is helpful in patients in whom there is suspicion of cauda equine syndrome, and when MSA is suspected (it is abnormal).

1. Urinary incontinence is defined as failure to store urine normally.
a. Stress urinary incontinence occurs when intra-abdominal pressure rises. It occurs because of weakened ligaments supporting the urethra (i.e., damage with childbirth) or because of a weak urinary sphincter due to lesions involving sacral neurons (Onuf’s nucleus).
b. Urinary urgency incontinence occurs when there is an overactive bladder resulting from decreased inhibition from the brain (“upper motor neuron bladder”). Incontinence occurs when bladder contraction pressure overcomes bladder neck outlet resistance. Urodynamic studies demonstrate detrusor overactivity (DO), with inappropriate bladder contractions during storage. Symptoms include frequency, nocturia, intermittent bladder contractions with incontinence, and urgency if sensation is preserved. Patients with urinary incontinence with urgency usually have upper motor neuron signs at neurologic examination. There are many causes: dementias, PD, hydrocephalus, bilateral frontal lobe lesion, spinal cord disease, syphilis, and tethered cord. In the elderly, there may be impaired cortical ability to inhibit the voiding reflex with urge incontinence. The non-neurologic cause of urgency is usually cystitis secondary to infection or inflammation with another cause.
2. Voiding difficulties with urinary retention result from an underactive detrusor (atonic bladder and bladder weakness), manifesting as a “lower motor neuron bladder.” Urodynamic study confirms a large, low-pressure bladder, and absent contractions. It is due to lesions affecting peripheral nerves or S2–S4 neurons innervating the bladder (acute upper neuron lesions can also cause detrusor weakness). There may be lack of awareness of bladder filling and inability to initiate voiding, producing urinary retention with overflow incontinence. Causes include spinal shock, myelitis, conus medullaris and cauda equina lesions, and neuropathy. Other causes are MSA, Friedreich’s ataxia, tabes, diabetes, alcoholic neuropathy, plexopathy, and after pelvic radiation.
3. Urgency incontinence and difficulty voiding with incomplete bladder emptying. Lesions in both the storage-facilitating areas of the brain (basal ganglia and pontine storage center) and the voiding-facilitating areas (PMC and sacral preganglionic intermediolateral cell column neurons) result in a combination of DO in the filling phase and underactive detrusor in the voiding phase, accompanied by urgency incontinence and difficulty voiding with incomplete emptying (detrusor hyperactivity with impaired contractile function—DHIC). DHIC is typical of MSA.
E. Treatment.
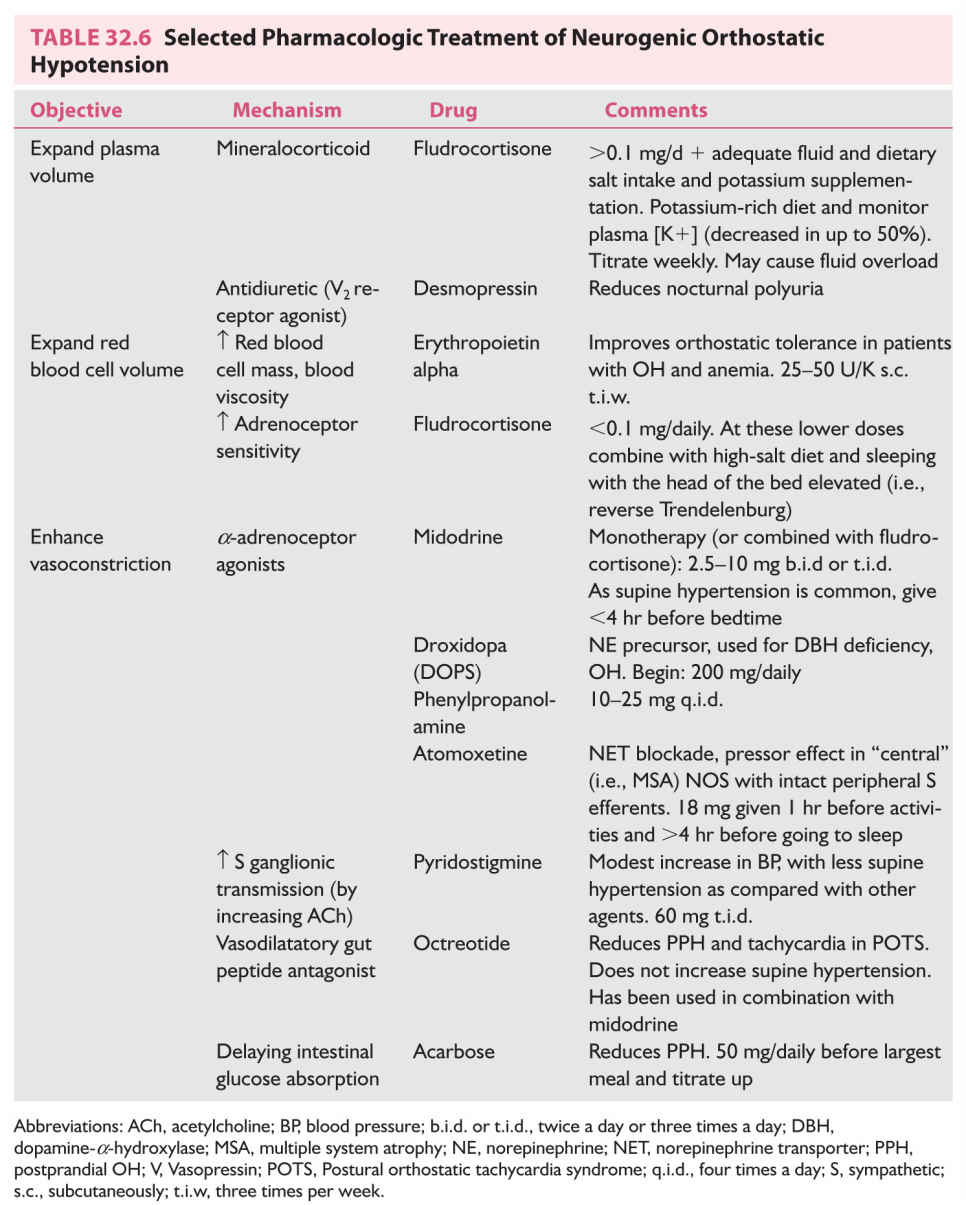
1. Bladder training. Timed bladder emptying, intermittent catheterization, and biofeedback techniques are helpful. Pelvic floor muscle strengthening with training exercises lead to decreased detrusor contractions and increased internal sphincter tone, with improvement of urinary frequency.
2. Bladder catheterization.
3. Pharmacotherapy for urinary storage dysfunction (Table 32.8). Injection of botulinum toxin type A has been used when there is no response to antimuscarinic drugs.
a. Urologic consultation is recommended.
GASTROINTESTINAL DYSFUNCTION
Esophageal, Gastric, and Intestinal Dysfunction
The ENS reflex pathways organized as myenteric (Auerbach’s) and submucosal (Meissner’s) plexus regulate gut motility, secretion and absorption, and vascular tone. ENS function is modulated by PS and S efferents.
Symptoms
Depending on the GI tract site affected, GI dysfunction manifests as esophageal dysphagia, gastroparesis, constipation, intestinal pseudo-obstruction, colonic inertia, megacolon, and defecatory dysfunction. Symptoms range from dysphagia, early satiety, fullness, bloating, nausea, postprandial vomiting, diarrhea, constipation, and fecal incontinence.
Causes
A. Drugs: opioids, tricyclic antidepressants, and dopamine agonists are frequent causes.
B. Mechanical obstruction and gut inflammation.
C. Neurologic: PD, MSA, spinal cord lesions, neuropathies (i.e., diabetic, amyloid), autoimmune ganglionopathies (paraneoplastic and primary), neuromuscular transmission defects, and myopathy are notable causes.
D. Other: Chagas disease is a common cause of autonomic GI dysfunction and should be considered with exposure to Trypanosoma cruzi-endemic areas. Rare causes include inherited neuropathies.
Fecal Incontinence
Fecal incontinence is associated with aging, reflecting a decline in muscle strength, mobility, and cognitive functioning. Typically there is loss of the sphincter’s endovascular cushion, thickening of external and internal sphincters, loss of resting and squeeze sphincter pressures, and reduced rectal sensation. Pudendal nerve stretch injury and direct trauma to the sphincter may produce pelvic floor dysfunction and fecal incontinence.
Typical symptoms range from insensible loss to sensory urge fecal loss, including complaints limited to perianal irritation.
Upper motor neuron lesions rostral to the sacral cord produce increased anal sphincter tone, loss of voluntary control, inability to relax or contract the sphincter on command, and result in fecal retention. With lesions of the sacral cord, conus medullaris, or cauda equina, there is a weak and areflexic anal sphincter with a patulous anus. Sensory loss may be present. Pelvic nerve lesions produce incontinence with loss of formed stool (diarrheal) with denervated external and internal anal sphincters.
Evaluation of Gastrointestinal Dysfunction
1. Physical examination with inspection of perineum, rectum, and vagina.
2. Direct observation via videoendoscopy.
3. Imaging studies: Dynamic imaging includes video-fluoroscopy, barium swallow studies, radionuclide gastric emptying studies, breath tests using radiolabeled substances (duodenal transit), colonic transit (markers, radionuclide), and defecography (barium, dynamic MRI). Anatomical imaging includes MRI of the spine (spinal cord lesions), pelvic computed tomography and/or MRI (malformations and structural abnormalities), endoanal ultrasonography (anal canal musculature and sphincter defects), and radiographic measurement of the rectoanal angle.
4. Manometry assesses GI sphincter function by measuring pressures.
5. Neurophysiologic studies. EMG of anal sphincter and puborectalis muscle and pudendal nerve terminal motor latency may help determine the type of neurologic disorder causing fecal dysfunction.
Management of Gastrointestinal Dysfunction
Gastroparesis
1. Diet with low-fat, low-fiber (to prevent bezoar), small and frequent meals
2. Prokinetic drugs: metoclopramide (also antiemetic), erythromycin
3. Pyloric sphincter botulinum
Constipation
1. Increase colonic content volume and maintain a near-normal consistency: high fiber content (>15 g/day), psyllium dietary fiber, methylcellulose, docusate sodium stool softener, lubiprostone (produces Cl-rich fluid secretion).
2. Laxatives: magnesium salts, lactulose, polyethylene glycol, bisacodyl (stimulates colonic contraction and increases fluid and NaCl secretion)
3. To prevent bacterial overgrowth due to slowed GI transit: metronidazole, ciprofloxacin, doxycycline (antibiotics are given for 1 week each month)
Fecal Incontinence
1. Techniques to achieve orderly defecation. Straining with Valsalva’s maneuvers, pressing on the abdomen (for patients with some preserved rectal sensation and able to feel the urge to defecate), digital stimulation of the rectum (most effective after glycerin suppositories, and in the sitting position). Scheduled enemas
2. Biofeedback training with EMG
3. Neuromodulation: anterior sacral root stimulators
4. Surgery: Replacement sphincter and pelvic floor reconstruction.
HYPO– AND HYPERHIDROSIS
Sweating abnormalities are frequent ANS symptoms. Sweating disorders may be idiopathic or part of small-fiber neuropathies, ANS ganglionopathies (see section Anhidrosis), MSA, and many other disorders. In addition to the potential disruption of thermoregulation, sweating disorders may be a source of disabling social embarrassment (hyperhidrosis).
Sweating is mediated by S cholinergic postganglionic neurons activating M3 muscarinic receptors of the eccrine sweat gland (vasointestinal peptide is also released to produce vasodilatation enhancing sweat production). Sweating stimuli may be thermoregulatory or emotional. Thermoregulatory sweating produces cutaneous vasodilatation and sweating distributed over the face, trunk, and proximal limbs, whereas emotional sweating produces vasoconstriction and seating is distributed over the palms and soles.
The pattern of the sweating abnormalities helps determine a cause. Distal patterns occur with length-dependent neuropathies, and generalized patterns reflect widespread ANS dysfunction as with MSA, ganglionopathies, and the effect of offending medication. Segmental S innervation helps to localize a spinal cord lesion. S innervation to the face originates from the T1–T2 spinal cord, the upper limbs from T4–T8, the trunk from T4–T12, and the lower limbs from T10–T12. Thermoregulatory sweat testing (where sweat produces changes in the color of substances—typically starch/iodine applied to the skin) provides clear anatomical sweating maps. Silicone imprint (sweat droplets are counted) and other more sophisticated tests provide indication of sweat production. QSART permits discrimination between preganglionic and postganglionic lesions.
Anhidrosis
Typically it is distributed over the upper body, head, and face. It may be accompanied by compensatory hyperhidrosis in proximal unaffected areas. Anhidrosis accompanies multiple ANS disorders, including MSA, PAF, peripheral neuropathies, autoimmune ganglionopathies, and CNS lesions (i.e., contralateral hypohidrosis with stroke) and the effect of various drugs. Acquired idiopathic anhidrosis selectively affects sweating, likely due to immune-mediated disruption of muscarinic sweat gland receptors.
Treatment relies on avoiding situations or environments that raise body temperature (i.e., adjusting clothing, exercising in cool environments), maintaining optimal hydration, and avoiding medication with anticholinergic effects, caffeine, and alcohol.
Hyperhidrosis
Generalized hyperhidrosis is usually associated with disorders producing S overactivity (i.e., alcohol withdrawal, thyrotoxicosis, pheochromocytoma, hypoglycemia, voltage-gated potassium channel antibodies, baroreflex failure, and HSAN III). It may also result from the effect of malignancies (i.e., lymphomas) and infections (i.e., tuberculosis), altering the thermoregulatory sweating threshold. Focal hyperhidrosis is frequent, and produces excessive sweating with mental stress and physical activity. It involves axillae, palms, and soles, and may be severe enough to cause significant social embarrassment. There are multiple syndromes associated with hyperhidrosis, many beyond the scope of this chapter.
Symptomatic treatment includes avoiding triggering factors (i.e., anxiety), topical preparations with aluminum chloride, tap water iontophoresis (palms, soles, axillae), oral medication (antimuscarinic anticholinergic drugs, clonidine, and topiramate), local injection of botulinum toxin, and in selected situations sympathectomy.
AUTONOMIC DISORDERS
Primary Autonomic Failure
A. Alpha-synucleinopathies. The ANS is typically involved with intracellular α-synuclein inclusions. In central ANS disorders (i.e., MSA), they are distributed in the CNS, and in peripheral ANS disorders they are mainly postganglionic (i.e., in LBD and PD, LBD), or exclusively postganglionic (i.e., PAF).
1. PAF (previously progressive autonomic failure, idiopathic OH, or Bradbury–Eggleston syndrome) is a sporadic degenerative disorder of middle to late life, affecting men more often. The cardinal manifestation is OH, with variable urinary sphincter and ED and constipation. Supine NE is low, and fails to increase with upright postural challenge (despite symptomatic hypotension).
In PAF postganglionic efferent S and PS autonomic neurons are impaired. The pathology is similar to that of idiopathic PD and DLB, with Lewy bodies in the CNS, S and PS ganglia, and in pre- and postganglionic neurons. Abnormalities in spinal cord and peripheral nerves are more prominent in PAF, perhaps explaining clinical differences between these disorders. Occasionally PAF may progress clinically to PD with OH and DLB with OH, and may develop dementia.
The differential diagnosis includes other causes of NOH (peripheral neuropathies with ANS disease, PD, and MSA). With PAF there are no parkinsonian, cerebellar, corticospinal, sensory, or lower motor neuron features, and ganglionic AChR are absent (autoimmune autonomic ganglionopathy [AAG] may be clinically similar to PAF). The clinical diagnosis requires the absence of symptoms other than autonomic >5 years, to distinguish PAF from cases of early MSA with isolated OH. Progression is slow, and, despite prominent OH, is less disabling than MSA.
2. MSA is a progressive, adult-onset neurodegenerative synucleinopathy involving both autonomic and somatic nervous systems, causing autonomic cardiovascular, urinary, and anorectal dysfunction, parkinsonism, and ataxia, in any combination. Onset is typically in the sixth decade of life, and men are affected more frequently. Early urinary and anorectal and ED with an abnormal sphincter EMG (due to loss of neurons of the sacral nucleus of Onuf) is typical. In many patients, chronic OH precedes other neurologic involvement, making differentiation of MSA from PAF difficult (Table 32.9). Pathology demonstrates abnormal oligodendroglial filaments of α-synuclein (glial cytoplasmic inclusions) mainly in substantia nigra, striatum, locus ceruleus, pons, inferior olives, cerebellum, and spinal cord, with neuronal degeneration and gliosis at multiple sites within the brain and spinal cord, but no Lewy bodies.

MSA is classified as either MSA-P (parkinsonism) or MSA-C (cerebellar), depending on the presence of predominant parkinsonism or cerebellar ataxia. As some clinical features of MSA are shared with other chronic progressive disorders such as PD and PAF, the clinical diagnosis may be difficult.
a. Clinical features. Patients with MSA typically have OH, erectile and urinary dysfunction, hypohidrosis, early instability, rapid progression, abnormal postures, bulbar and respiratory dysfunction, and emotional incontinence and Parkinsonism and cerebellar features.
b. Laboratory evaluation.
(1) Autonomic testing (Table 32.3)
(2) EMG may suggest involvement of the anterior horn cells in MSA. Abnormal sphincter muscle EMG (denervation potentials of spinal cord anterior horn cells) can distinguish MSA from PD (Onuf’s neurons are spared) in the first 5 years after the onset of symptoms and signs, and from PAF, as well as from cerebellar ataxias, if other causes for sphincter denervation have been ruled out. Normal EMG is unlikely in MSA.
(3) MRI. Linear hyperintense putaminal border rim, putaminal atrophy, and putaminal hypointensity relative to the globus pallidus signal, and cruciform pontine hyperintensities (“hot cross bun” pattern) are specific to MSA, but sensitivity is low. Cerebellar atrophy may be present in some patients even without clinical cerebellar signs.
c. Management (Tables 32.5 and 32.6). Only symptomatic treatment is available. One-third of patients have temporary response to levodopa; some may respond to amantadine. Side effects, especially accentuation of hypotension, must be kept in mind.
AUTONOMIC NEUROPATHIES
Autonomic neuropathies are peripheral neuropathies where ANS fibers are prominently affected. Impairment of the lightly myelinated or unmyelinated ANS fibers may interfere with the normal functioning of many organ systems, leading to various cardiovascular, urogenital, GI, sudomotor, thermoregulatory, and pupillomotor autonomic symptoms. Injury and dysfunction of ANS innervation may be secondary to diabetes, alcoholism, infections, amyloidosis, connective tissue disorders, and paraneoplastic syndromes, or occur in isolation without evident underlying disease (primary autonomic neuropathies). Depending on the extent and location of an autonomic lesion, ANS neuropathy may be widespread or limited to neurotransmitter type (cholinergic and adrenergic neuropathies), an organ system, or to distal small fibers. Selected examples are discussed here.
Diabetic autonomic neuropathy (DAN), a chronic autonomic neuropathy, is a common complication of diabetes. DAN may involve cardiovascular, GI, urogenital, and thermoregulatory (sweating) functions. As a length-dependent neuropathy, DAN affects the vagus nerve early on with abnormal cardiovascular autonomic function, manifested as reduced HR variation, the earliest indicator of cardiac autonomic neuropathy (CAN). The 5-year mortality rate is five times higher for those with CAN than for individuals without cardiovascular autonomic involvement. Clinical symptoms of DAN generally do not occur until long after the onset of diabetes, and are more common with worse glycemic control. In patients with long-standing diabetes, esophageal transit is delayed in 50% and gastroparesis is present in 40%. Symptoms are variable and more common in patients with worse chronic glycemic control and with psychological disorders. Other common GI symptoms include constipation, diarrhea, and fecal incontinence. Impaired glucose regulation (impaired glucose tolerance [IGT], nondiabetic hyperglycemia, prediabetes) with small-fiber neuropathy is accompanied by mild autonomic neuropathy (sudomotor fibers tend to be affected earlier with IGT).
AAG is a primary acute and subacute autonomic neuropathy characterized by a rapid onset of isolated diffuse ANS dysfunction associated with ganglionic nicotinic a 3-acetylcholine receptors (a 3-AChR) autoantibodies. AAG may follow respiratory and GI infections, vaccines, surgery, treatment with interferon, and may be a paraneoplastic presentation (30% have malignancies, mostly adenocarcinoma). Typical features of the classic form of AAG are acute/subacute widespread and frequently severe involvement of the S, PS, and ENSs (GI dysmotility is prominent). It is a monophasic disorder with high (>0.5 nmol/L) antibody levels (ANS manifestations are more pronounced with higher levels). Other forms are slowly progressive and chronic, ANS involvement is more localized, and a 3-AChR antibody levels are low. As slowly progressive and chronic forms of ANS dysfunction (including PAF) may be due to AAG, these patients should be screened for AAG. Postural tachycardia, chronic idiopathic anhidrosis, isolated GI dysmotility, and distal small-fiber neuropathies may have low a 3-AChR antibody levels. Symptoms may improve with immunotherapy, although in acute and subacute forms improvement (usually partial) may also be spontaneous. Chronic forms of AAG should be treated early on in an attempt to prevent disease progression.
Paraneoplastic autonomic neuropathy is frequently present with paraneoplastic disorders. Anti-Hu, anti-CV2/CRMP-5, and a 3-AChR antibody are frequently associated with autonomic involvement (GI dysmotility is prominent). Various antineuronal (typically anti-Hu, anti-VGCC, and antiganglionic acetylcholine receptor) antibodies associated with underlying disease (usually a paraneoplastic syndrome associated with lung, thymus, gyn, and breast cancer) may be detected. Treatment of the underlying tumor is the main therapeutic approach. Immunomodulatory therapy can be beneficial in some cases
Enteric ganglionitis (EG) is an example of an autonomic disorder affecting the gut exclusively. It is characterized by inflammatory or immune dysfunction of intrinsic GI innervation causing dysmotility and delayed transit. Lymphoid infiltration of the small intestine produces intestinal pseudo-obstruction; infiltration of the myenteric ganglia causes achalasia. EG is usually associated with paraneoplastic neurologic syndromes, but may be also present with CNS disorders and with Chagas disease.
AUTONOMIC CRISIS
A. Autonomic crises. Acute autonomic dysfunction occurs in many conditions and a hypersympathetic state is most often encountered. Examples of such neurologic conditions are as follows.
1. Cerebral lesions. Ischemic stroke, intracerebral hemorrhage, subarachnoid hemorrhage, intracranial mass lesion, Cushing’s response
2. Spinal cord lesions (in particular with an injury at level T6 or above)
3. Peripheral nerve disease. Guillain–Barré syndrome (GBS)
4. Systemic diseases. Tetanus, episode of acute intermittent porphyria
5. Drug-related conditions. Neuroleptic malignant syndrome, sympathomimetic drug overdose, tricyclic antidepressant overdose
B. Autonomic dysreflexia is a life-threatening “sympathetic storm” that occurs in cases of spinal cord transection. The spinal cord lesion is usually above the midthoracic level. The episodes are paroxysmal and start several months after the acute spinal cord injury as recovery occurs. They are characterized by sudden onset of severe hypertension, headache, sweating and flushing, piloerection, and sometimes chills due to uninhibited activity of spinal S neurons. A precipitating cause can often be identified, and is usually a noxious stimulus. Urinary bladder distention, constipation, and fecal impaction are common causes. Elimination of the precipitating cause often results in resolution of the episode, and prevention is the best therapy. Hypertension is treated with antihypertensive agents with rapid onset and short-duration effects.
C. Anesthetic complications. Patients with autonomic dysfunction may have unpredictable, life-threatening, intraoperative hypotension and cardiovascular collapse. Caution and close operative and postoperative observation and monitoring are necessary, and including with spinal anesthesia may result in hypotension because of volume shifts (it may produce further loss of autonomic control of splanchic bed volumes).
Mechanisms
A. Impaired cardiovascular reflexes with inadequate compensation for anesthetic-induced vasodilatation and volume shifts, unpredictable responses to vasoactive agents due to denervation supersensitivity, and impaired response to atropine (due to cardiac PS denervation, as in DAN)
B. Impaired central respiratory reflexes with hypopnea and apnea
C. GI autonomic dysfunction with gastroparesis and increased risk for aspiration
D. Impaired temperature regulation with hyperthermia (due to impaired sweating) or hypothermia (due to deficient vasoconstriction)
E. Decreased hepatic clearance of drugs due to liver hypoperfusion during hypotension
Prevention
Prevention includes:
1. Optimal hydration and BP management
2. Rapid sequence induction of anesthesia and volatile anesthetics in high-risk patients with autonomic dysfunction
3. Caution and close operative and postoperative observation and monitoring. Particular attention is given when switching ventilation parameters, as this may precipitate acute hypotension because of changes in venous return. Hyperventilation should be avoided as it aggravates hypotension.
Key Points
• The ANS maintains the body’s internal homeostasis and regulates its protective responses.
• Symptoms of ANS dysfunction reflect the widespread ANS innervation, and their differential diagnosis is broad.
• The site of an autonomic lesion, the clinical course, and the presence or absence of accompanying somatic neurologic and/or systemic or localized manifestations of disease help establish a cause of autonomic dysfunction.
• NOH is a frequent and disabling manifestation of ANS dysfunction.
• Nonpharmacologic management of symptomatic OH is key to successful treatment.
• ANS dysfunction may be serious and life threatening in certain instances, and these should be sought and managed appropriately.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


