Autosomal Dominant Cortical Myoclonus and Epilepsy (ADCME) with Linkage to Chromosome 2p11.1-q12.2
Renzo Guerrini*
Lucio Parmeggiani*
Carla Marini*
Paola Brovedani*
Paolo Bonanni*
*Epilepsy, Neurophysiology and Neurogenetics Unit, Department of Child Neurology and Psychiatry, University of Pisa and Research Institute ‘Stella Maris’ Foundation, Pisa, Italy
Introduction
Autosomal dominant cortical myoclonus and epilepsy (ADCME) is a recently characterized nonprogressive familial syndrome that combines rhythmic cortical reflex myoclonus with focal and generalized seizures (1). In this chapter, we outline the clinical and neurophysiologic features that characterize ADCME and help to differentiate it from other conditions with similar features, such as familial adult myoclonic epilepsy (FAME).
ADCME patients present with a complex of symptoms forming a homogeneous syndromic core (Fig. 21-1), including semicontinuous rhythmic distal jerking (also termed cortical tremor), which is a particular form of cortical reflex myoclonus, generalized tonic–clonic seizures (GTCSs) that are occasionally preceded by generalized myoclonic jerks, and both generalized and focal electroencephalogram (EEG) abnormalities. Age at onset of cortical tremor and of GTCSs overlap in a given individual but vary between individuals, ranging from 12 to 50 years (1). Some patients also present with focal, intractable seizures. The pattern of inheritance is autosomal dominant with high penetrance and linkage to chromosome 2p11.1-q12.2.
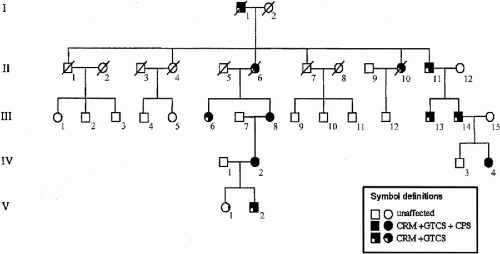 FIG. 21-1. ADCME family pedigree. CPS, complex partial seizures; CRM, cortical reflex myoclonus; GTCS, generalized tonic–clonic seizures. |
Clinical and Genetic Features
Clinical Neurophysiology of Cortical Tremor
The distal jerking observed in patients with ADCME has the neurophysiologic characteristics of cortical tremor (2,3). A stereotyped pattern of rhythmic, involuntary hand movements is evident during isometric muscle contraction and is accompanied by rhythmic EMG bursts at 8 to 15 Hz, involving synchronously agonist and antagonist muscles of the forearms (Fig. 21-2). Involvement of more proximal as well as facial muscles, especially the eyelids, is possible. Backaveraging and frequency analysis indicate that EEG activity is coherent with electromyography (EMG) at the frequency of the myoclonus and that time-locked EEG activity precedes EMG bursts (1,4) (Fig. 21-2). The C-reflex at rest is enhanced, somatosensory-evoked potentials (SEPs) are of high amplitude (Fig. 21-3), motor threshold at rest, obtained by transcranial magnetic stimulation, is reduced, and the silent period following motor-evoked potentials (MEP) is shortened (Fig. 21-2). Clinical and neurophysiologic characteristics suggest that myoclonic activity, in addition to a high rhythmicity, also has propensity for intra- and interhemispheric cortical spread. Intermittent photic stimulation (IPS) at low rates evokes single occipital spikes in response to each flash and pattern-visual-evoked potentials (P-VEPs) are of very high amplitude (Fig. 21-3).
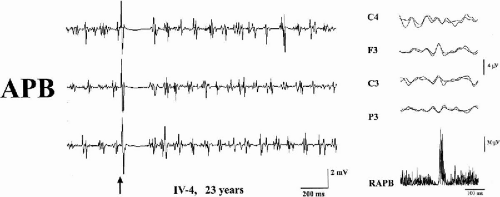 FIG. 21-2. Left: Rhythmic EMG bursts at a frequency of around 15 Hz are observed during slight muscle contraction. Transcranial magnetic stimulation administered at the time, indicated by the arrow, is followed after around 20 ms by a motor-evoked potential (MEP) that is followed by a post-MEP silent period. Rhythmic EMG bursts reappear soon after the post-MEP silent period, supporting a cortical hyperexcitability. The three traces represent three different replications. Right: Backaveraging from the onset of 100 rhythmic EMG bursts. A positive–negative potential, recognizable over the contralateral frontocentral electrodes, precedes the jerk by about 15–20 ms. Two traces are overlapped to show data reproducibility. RAPB, right abductor pollicis brevis. |
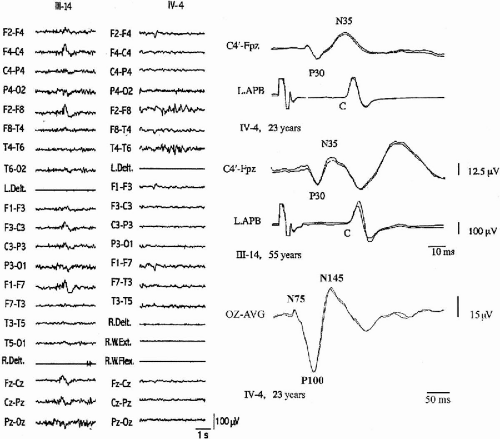 FIG. 21-3. Left: Interictal EEG–EMG recordings during relaxed wakefulness in Patients IV-4 and III-14. Interictal discharges show equipotentiality over the right frontotemporal leads. Right, top and middle: Cortical SEPs to electrical stimulation of the left median nerve at the wrist are recorded in the same patients whose EEG recordings are presented on the left. An enlarged N20–P30–N35 complex on right centroparietal region is followed by C-reflex in the left abductor pollicis brevis. The shock-C-reflex latency is ~45 msec. Right, lower: VEP in response to checkerboard stimulation in patient IV-4. An enlarged N75-P100-N145 complex is recorded. APB, abductor pollicis brevis; C, cortical reflex; Delt., deltoid muscle; Ext, extensor; Flex, flexor; L, left; R, right; W, wrist. |
These combined findings suggest widespread cortical hyperexcitability with deficits in inhibitory cortical mechanisms (5,6,7,8,9). The prime origin of rhythmic motor cortex excitation is difficult to demonstrate. Rhythmic generators within cortical layers IV and V (10) might play a role. Alternatively, cortical neurons could be driven by a subcortical generator (3). The corticomuscular loop underlying cortical reflex myoclonus in a highly excitable sensorimotor cortex may also play a role. Each afferent volley generated by a myoclonic jerk might, in turn, facilitate an efferent volley that sustains an ensuing myoclonic jerk, thereby sustaining a rhythmic afferent–efferent mechanism.
Cortical tremor may be observed in different clinical settings, as a consequence of multiple etiologic factors enhancing motor cortex excitability. In addition to FAME (2,9,10,11,12), it may also be observed in other genetic disorders, including a form of familial mental retardation and generalized epilepsy (13), Angelman syndrome (7), Rett syndrome (personal observation), and progressive myoclonus epilepsies (PME) (3), in which it may be accompanied by generalized as well as focal seizures. In addition, cortical tremor has been observed in association with focal motor seizures after ischemic brain lesions involving the sensorimotor cortex (16,17), in the presence of cortical tubers in tuberous sclerosis (18) or, as an isolated symptom after surgical removal of a frontal lobe meningioma (19), and in patients with no other neurologic abnormality (3). These observations indicate that cortical tremor is a relatively common neurologic manifestation and is often associated with epilepsy of either symptomatic or genetic etiology. It is, therefore, likely that the motor cortex has an intrinsic propensity for rhythmicity that is uncovered by different etiologic conditions having in common the ability to lower cortical inhibition, either regionally or more diffusely.
Related posts:
 Progressive Myoclonus Epilepsies: EPM1, EPM2A, EPM2B
Progressive Myoclonus Epilepsies: EPM1, EPM2A, EPM2B
 Myoclonic Status in Nonprogressive Encephalopathies
Myoclonic Status in Nonprogressive Encephalopathies
 Myoclonic–Astatic Epilepsy of Early Childhood – Definition, Course, Nosography, and Genetics
Genetics of Juvenile Myoclonic Epilepsy: Faulty Components and Faulty Wiring?
Ketogenic Diet in Patients with Dravet Syndrome and Myoclonic Epilepsies in Infancy and Early Childhood
Myoclonic–Astatic Epilepsy of Early Childhood – Definition, Course, Nosography, and Genetics
Genetics of Juvenile Myoclonic Epilepsy: Faulty Components and Faulty Wiring?
Ketogenic Diet in Patients with Dravet Syndrome and Myoclonic Epilepsies in Infancy and Early Childhood
 Treatment of Myoclonic Epilepsies of Childhood, Adolescence, and Adulthood
Treatment of Myoclonic Epilepsies of Childhood, Adolescence, and Adulthood
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


