Basal Ganglia
Anatomy of the Basal Ganglia
Basal ganglia (Fig. 19.1) include the corpus striatum, the substantia nigra (pars compacta and a pars reticularis), the subthalamic nucleus of Luys, and the ventral tegmental area. The corpus striatum comprises the striatum proper (or neostriatum), made up of the putamen, caudate nucleus, and nucleus accumbens, and the globus pallidus (or paleostriatum), with its medial or internal (Gpi) and lateral or external (Gpe) segments and the ventral pallidum, with its internal and external portions.
The basal ganglia play a major role in the control of posture and movement. Marsden [241,242] proposed that “the basal ganglia are responsible for the automatic execution of learned motor plans;” that is, the basal ganglia mediate movements that have been “laid down by practice” and that are subconscious. Examples of such movements include shifting in bed and walking, as well as the proximal movements that support distal ones. Someone typing a letter may be conscious of the movements of the fingers on the keyboard but is not generally conscious of the shoulder abduction and extension that allow the fingers to fly over the keyboard.
The major anatomic connections of the basal ganglia [6,48] are complex (Fig. 19.2) and include several “closed circuits” of connections [277,287,294]. In essence, the basal ganglia consist of an input zone, comprising the putamen, caudate nucleus, and ventral striatum, and an output zone, comprising the medial globus pallidus and the substantia nigra pars reticularis. The main outputs from the medial globus pallidus and substantia nigra pars reticulata are to the thalamus, and thence to the premotor (e.g., supplementary motor area, anterior cingulate motor area, and lateral premotor cortex) and frontal lobe structures. Some of the main connections are discussed in subsequent text.
Inputs into the Striatum (Caudate and Putamen)
CORTICAL PROJECTIONS TO THE NEOSTRIATUM
All parts of the cerebral cortex give rise to efferent fibers to the caudate and putamen. These corticostriate projections terminate mainly ipsilaterally in a topographic pattern (e.g., the frontal cortex projects fibers to the ventral head of the caudate and rostral putamen). The cortex also sends fibers to the substantia nigra, subthalamic nucleus, and claustrum.
THALAMOSTRIATAL PROJECTIONS
The intralaminar nuclei of the thalamus, especially the centrum medianum (CM) nucleus, send fibers to the striatum.
NIGROSTRIATAL PROJECTIONS
Fibers originating in the pars compacta of the substantia nigra project to the striatum (caudate nucleus, putamen, and globus pallidus).
RAPHE NUCLEI-STRIATAL PROJECTIONS
The brainstem raphe nuclei send ascending fibers to the striatum.
Striatal Efferents
Most striatal efferents project to the globus pallidus (to both the internal and external segments). Other striatal efferents go to the substantia nigra.
Pallidal Afferents and Efferents
The globus pallidus receives ascending afferent fibers from the substantia nigra and subthalamus (mainly to the medial or internal pallidum). Both the external and internal globus pallidus also receive afferents from the striatum.
The major outflow from the globus pallidus arises from the internal portion and projects to the ventral anterior (VA) and ventral lateral (VL) nuclei of the thalamus. These thalamic nuclei also receive afferents from the pars reticularis of the substantia nigra. Because the VL thalamic nucleus projects to the motor cortex and the VA thalamic nucleus projects to the premotor cortex, the major basal ganglia efferents influence the motor system.
Efferents from the internal globus pallidus also project to the CM thalamic nuclei, which in turn project to the putamen. A closed circuit is thereby formed: putamen—internal pallidum—central medianum nucleus—putamen. The internal globus pallidus also sends fibers to the lateral habenular nucleus.
The external portion of the pallidum sends fibers to the internal pallidum and to the subthalamic nucleus. The subthalamic nucleus in turn sends fibers to both the internal and external pallidum. Therefore, another closed circuit is formed: external globus pallidus—subthalamus—external and internal globus pallidus.
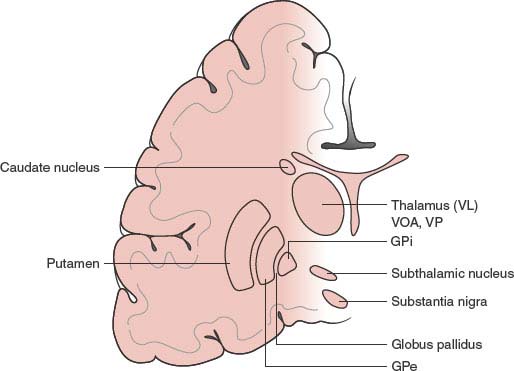
FIG. 19.1. Anatomy of the basal ganglia. VL = Ventrolateral thalamic nucleus; VA = ventral anterior thalamic nucleus; VP = ventroposterior thalamic nucleus; GPi = globus pallidus pars interna; GPe = globus pallidus pars externa.
Other pallidal efferents also project to the substantia nigra, red nucleus, inferior olive, hypothalamus, and mesencephalic reticular formation.
Nigral Afferents and Efferents
The pars reticularis of the substantia nigra receives fibers from the cerebral cortex, the striatum, the globus pallidus, and the subthalamic nucleus. Pars reticularis efferents project to the VA and VL thalamic nuclei and to the reticular formation and superior colliculus.
The pars compacta of the substantia nigra sends dopaminergic fibers to the caudate nucleus and putamen. This output is excitatory for the striatal neurons of the direct pathway and inhibitory to the striatal neurons of the indirect pathway, described in the subsequent text.
It can thus be seen that the basal ganglia exert their influence mainly by way of the cerebral cortex (i.e., they do not send fibers that connect directly with brainstem and spinal cord structures). They provide a subcortical network by which the entire cerebral cortex can influence the motor system (motor and premotor cortex), mainly by the following circuit: cerebral cortex—neostriatum—globus pallidus and substantia nigra—VA and VL thalamic nuclei-motor and premotor cortex.
Both anatomically and physiologically, a direct and an indirect system have been described in the striato-pallido-thalamic projection. In the direct system, the putamen and the caudate receive excitatory input from the pars compacta of the substantia nigra and project inhibitory fibers (g-aminobutyric acid, or GABA, colocalized with substance P) to the medial globus pallidus and to the pars reticularis of the substantia nigra, which in turn, inhibit the ventrolateral nucleus of the thalamus. Stimulation of this system would disinhibit the ventrolateral nucleus of the thalamus, resulting in cortical excitation. In the indirect system, the putamen and caudate receive inhibitory input from the pars compacta of the substantia nigra and project inhibitory fibers (GABA colocalized with enkephalin) to the lateral globus pallidus which, in turn, inhibits (through GABA) the subthalamic nucleus. The subthalamic nucleus stimulates (through glutamate) the medial globus pallidus, inhibitory over the ventrolateral nucleus of the thalamus. Stimulation of this system inhibits the ventrolateral nucleus of the thalamus and results in cortical inhibition.
In functional terms, the activities of the basal ganglia pathways within the striatopallidal motor loop can, therefore, be summarized as follows:
1. Corticostriatal input from the sensorimotor cortex (glutamate) or input from the substantia nigra pars compacta (dopamine) excite direct inhibitory pathways of the putamen and medial globus pallidus and substantia nigra pars reticulata resulting in disinhibition of thalamic nuclei, which project excitatory input to the precentral motor fields. The net effect is facilitation of cortically initiated movement.
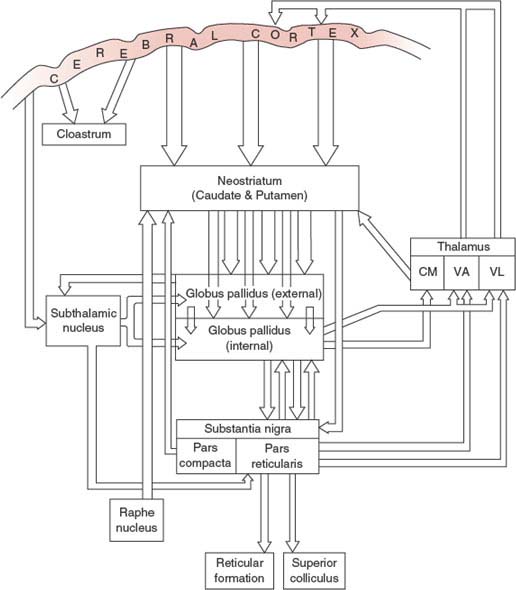
FIG. 19.2. Anatomic connections of the basal ganglia. CM = centromedian thalamic nucleus; VA = ventral anterior thalamic nucleus; VL = ventrolateral thalamic nucleus.
2. Corticostriatal stimulation of the indirect system has a net effect of releasing subthalamic input to the medial globus pallidus, leading to increased inhibition of thalamic nuclei and reduced thalamo-cortical input to the central motor cortex. Dopamine nigro-striatal input inhibits this indirect pathway, thereby reducing negative feedback to the precentral motor system.
3. Deprivation of the dopamine nigro-striatal input reduces the positive feedback to the precentral motor system through the direct striatopallidal system and increases the negative feedback through the indirect system. The overall effect of dopamine depletion in the striatum would be the inhibition of the cortically initiated movement, whereas overactivity of the dopamine in the striatum would stimulate the cortically initiated movement.
Although helpful in conceptualizing most of the anatomo-physiologic data regarding the connectivity of the basal ganglia, this currently accepted schema does not explain some clinical findings in basal ganglia diseases [244].
Lesions of the Basal Ganglia
Pathologic processes affecting the basal ganglia are often diffuse. When discrete, they usually also affect neighboring structures, such as the internal capsule, the hypothalamus, and the white matter of the cerebral hemispheres. Therefore, except for hemiballismus often associated with damage to the contralateral subthalamic nucleus, correlation between basal ganglia lesions and clinical motor dysfunction tends to be obscure.
The literature concerning behavioral effects of lesions of the basal ganglia in experimental animals is often conflicting, and these lesions rarely produce models of human movement disorders. Therefore, these studies are not reviewed. In general, stimulation and destructive lesions of the caudate, putamen, and globus pallidus produce inhibition of movement or contralateral body turning [252].
Some disorders in humans associated with lesions of the basal ganglia are as follows:
1. Lesions of the subthalamic nucleus produce contralateral hemiballismus [59].
2. Small unilateral lesions of the anteroventral portion of the caudate cause contralateral choreoathetosis [217].
3. Unilateral lesions of the globus pallidus may cause contralateral hemidystonia, hemiparkinsonism, or tremor, whereas bilateral globus pallidus lesions may cause dystonia, parkinsonism, abulia, or akinesia [267].
4. Lesions of the substantia nigra result in parkinsonism.
5. Unilateral basal ganglia (pallidal-putaminal) hemorrhages or lacunar infarcts may present with sudden falling to the contralateral side while sitting, standing, or walking [203]. The falls are distinctly slow, tilting motions in a stereotyped lateral or diagonal trajectory (“like a falling log”) and occur with the eyes open but are exacerbated by eye closure.
In a study of behavioral and movement disorders with lesions affecting the basal ganglia, lesions of the caudate nucleus rarely caused motor disorders (e.g., chorea or dystonia) but were more likely to cause behavioral problems, especially abulia (apathy with loss of initiative and of spontaneous thought and emotional responses) or disinhibition [37]. Lesions of the putamen and globus pallidus rarely caused abulia and did not produce disinhibition but commonly caused dystonia, particularly when the putamen was involved. Bilateral lesions of either the putamen or the globus pallidus caused parkinsonism or dystonia-parkinsonism infrequently. The prominence of behavioral disturbances with caudate lesions emphasizes the more complex cognitive role of this structure, whereas the frequent occurrence of dystonia and less commonly parkinsonism with lesions of the putamen and globus pallidus emphasizes the motor roles of these structures [37]. In another study of patients with lenticular infarcts, two distinct clinical syndromes corresponding to the two anatomical areas of the lenticular nucleus were described [138]. Infarcts within the globus pallidus were associated with behavioral and cognitive disorders, whereas infarcts in the putamen caused motor disorders (dystonia) and cognitive impairment. Also, damage to a frontal-caudate functional system likely underlies the aphasias (language disturbances) resulting from subcortical lesions affecting the deep frontal and paraventricular white matter (subcortical aphasias) [258].
Movement disorders can be defined as neurologic dysfunctions in which there is either an excess of movement (abnormal involuntary movements, or AIMs; hyperkinesias; dyskinesias) or a paucity of voluntary and automatic movements (akinesia, bradykinesia, or hypokinesia) unassociated with weakness or spasticity. Paucity of movement characterizes the disorder known as parkinsonism. The dyskinesias are discussed first.
Dyskinesias
Chorea
Chorea [108,327] is characterized by sudden, brief, spontaneous, involuntary, purposeless, continuous, irregular, and unpredictable jerks that randomly involve the appendicular, facial, or truncal musculature. The muscles involved vary depending on the underlying disease process (e.g., truncal involvement predominates in Huntington chorea and distal appendicular involvement is predominant in Sydenham chorea). The hyperkinetic movements may be unilateral (hemichorea) or bilateral, occur at rest or during volitional acts, interfere with activities of daily living, cease during sleep, intensify during stress, and are often camouflaged by the patient through a superimposed purposeful act (parakinesia).
Chorea is often associated with changing muscle tone (e.g., hypotonia in Sydenham chorea, rigidity and hypokinesis in the Westphal variant of Huntington disease, and dystonia in the juvenile variant of Huntington disease). Patients with chorea tend to hyperpronate the upper extremity when attempting to maintain an extended posture, and they are often unable to sustain a tight handgrip (milkmaid’s grip). The tongue cannot be maintained in a protruded position and darts in and out irregularly (trombone or flycatcher tongue). Facial grimacing and abnormal respiratory sounds may be manifestations of chorea.
Huntington disease is an autosomal dominant disorder with the defect localized to the short arm of chromosome 4. The mutant gene contains an expansion of CAG trinucleotide repeats that code for a protein huntingtin. The longer the repeat length (normal is 10–29 copies), the earlier the onset of the disease. Symptoms often begin insidiously in the third through the fifth decades and are characterized by progressive chorea, dystonia, eye movement abnormalities, behavioral changes, and progressive dementia. The choreatic movements may antedate or follow the dementia. Saccadic eye movements become slow and uncoordinated, and smooth pursuit eye movements are frequently disrupted by saccadic intrusions. An increased rate of suicide has been reported in patients with Huntington’s disease. Huntington’s disease-like 2 (HDL2) is a progressive autosomal dominant disease with marked clinical and pathologic similarity to Huntington disease [237,355]. The gene defect is a CTG/CAG expansion in a variably spliced exon of the junctophilin-3 gene. HDL2 can present with acanthocytosis, weight loss, and incoordination usually in the fourth decade. Patients may develop dystonia, chorea, rigidity, dysarthria, hyperreflexia, bradykinesia, tremor, psychiatric symptoms, and dementia. Other examples of autosomal dominant disorders associated with chorea include benign hereditary chorea (BHC) (onset in childhood), spinocerebellar ataxia 3 (SCA 3 or Machado-Joseph disease), and dentatorubropallidoluysian atrophy (DRPLA).
Benign hereditary chorea (BHC) is an autosomal dominant disorder often presenting with childhood-onset chorea, no dementia, and little or no progression [116,186]. In some patients with infancy-onset BHC, treatment with levodopa improves gait abnormalities and chorea [19]. Another hereditary (autosomal recessive) cause of chorea is familial degeneration of the basal ganglia with acanthocytosis [39]. Less common causes of hereditary chorea include Wilson disease, Lesch-Nyhan syndrome, dopa-responsive dystonia (Segawa syndrome), Niemann-Pick disease, Pelizeus-Merzbacher disease (sudanophilic leukodystrophy), Hallervorden-Spatz disease (pantothenate kinase-associated neurodegeneration or PKAN), ataxia telangiectasia, Leigh syndrome (subacute necrotizing encephalomyelopathy), tuberous sclerosis, mitochondrial encephalopathies, glutaric academia type 1, GM1 and GM2 gangliosidoses, neuroacanthocytosis, neuroferritinopathy, and the hereditary forms of paroxysmal choreoathetosis.
Neuroacanthocytosis (chorea-acanthocytosis)(NA) is a familial or nonfamilial multisystem progressive disorder in which chorea occurs in almost all cases [39,150]. The mean age of onset is 32 years (range 8–62 years) and patients demonstrate acanthocytosis with normal lipoproteins. Initial lip and tongue biting are followed by orolingual “eating” dystonia, motor and phonic tics, generalized chorea, akinetic-rigid features, vertical ophthalmoparesis, and seizures [23]. The orofacial-lingual involuntary dystonic movements and pseudo-bulbar disturbance commonly cause dysphagia and dysarthria. Cognitive impairment, psychiatric features, and organic personality changes are common. Other signs and symptoms include amyotrophy, axonal neuropathy, decreased or absent muscle stretch reflexes, and increased serum creatine kinase (CK) without myopathy [150]. The abnormal gene found in neuroacanthocytosis, on chromosome 9, is an evolutionarily conserved protein called chorein that is probably involved in protein sorting. Chorea may also be associated with neurogenic atrophy in cases of Fotopoulos syndrome [285].
Walker et al. reviewed the clinical and laboratory features of NA [354]. NA may be divided into three groups: (a) the “core” NA syndromes with neurodegeneration of the basal ganglia, comprising autosomal recessive chorea–acanthocytosis (ChAc) due to mutation of VPS13 A and X-linked McLeod syndrome (MLS) due to mutation of the XK gene on the X chromosome; (b) conditions with decreased lipoproteins, namely, abetalipoproteinemia (Bassen–Kornzweig syndrome) and hypobetalipoproteinemia, in which the hallmarks are peripheral neuropathy and ataxia due to dorsal column degeneration, but movement disorders are not present; and (c) conditions in which acanthocytosis is occasionally seen, such as PKAN and HDL2. In addition, acanthocytosis has been reported in mitochondrial disease. Chorea is the clinical hallmark of ChAc, MLS, HDL2, and PKAN, but a variety of movement disorders may be seen, including dystonia, less frequently parkinsonism, and rarely tourettism. In “atypical” cases of PKAN, less likely to be due to mutations of PKAN2, rigidity is more prominent. Severe orofacial dystonias with tongue and lip biting are typical of ChAc and are less suggestive of MLS or the other disorders. The tongue thrusting can cause marked problems with eating. Speech difficulties, specifically palilalia or dysarthria, are prominent features of PKAN. Patients with ChAc, MLS, or HDL2 often present with neuropsychiatric symptoms, prior to development of the movement disorder. Psychiatric symptoms may include bipolar disorder, schizoaffective disorder, obsessive-compulsive disorder, depression, anxiety, and agitation. Cognitive problems may range from minor abnormalities to dementia, with deficits primarily in memory and executive skills. Children with PKAN, especially the atypical form, may develop cognitive decline and various psychiatric abnormalities, and occasionally present with these features. Seizures are seen in 40% of patients with ChAc and 50% of patients with MLS. In the French Canadian ChAc population, this number is much higher, with temporal lobe seizures often being the presenting feature, several years before the appearance of the movement disorder. Seizures have not been reported in HDL2 or PKAN. Oculographic studies in ChAc have revealed slowed and hypometric saccades and frequent square-wave jerks and oscillations, similar to those seen in Huntington disease. The majority of patients with typical PKAN develop retinopathy, and various eye movement abnormalities have been documented, including saccadic pursuit, impaired saccades, abnormal vestibulo-ocular reflex, and pupillary responses. Autonomic nervous system dysfunction in ChAc may result in abnormal respiratory rhythm, orthostatic hypotension, and impaired digestive motility. Autonomic dysfunction has been reported in MLS with hypersalivation and excessive sweating. Cardiomyopathy and arrhythmias are observed in approximately 60% of MLS patients but are not typical for ChAc. Childhood onset, at ages younger than 10 years, is seen in typical cases with the PANK2 mutation. Presentation with atypical features and a slower disease progression may be seen in young adults. ChAc usually manifests clinically between ages 20 and 40 and rarely below age 20 or after 50. The youngest reported age was 16 years. MLS typically develops in men aged 40 to 60. Exceptionally, female mutation carriers manifest the disease. The age at onset of HDL2 is variable. As in HD, longer trinucleotide repeat expansions correlate with a younger age at onset. The minimum repeat expansion size for disease expression is reported to be 40, although a subject with 43 repeats appeared to be asymptomatic at age 65. All patients reported to date have been of African ancestry.
Neuroferritinopathy is a dominantly inherited, progressive, potentially treatable movement disorder caused by mutation of the ferritin light chain gene (FTL1) [70,81,263]. Patients may have low serum ferritin levels and brain histochemistry shows abnormal aggregates of ferritin and iron. Patients with this disorder present with variable symptoms, beginning in the third to sixth decade, including chorea, dystonia, or an akinetic-rigid syndrome. Features overlap with common extrapyramidal disorders: idiopathic torsion dystonia, idiopathic Parkinson disease and Huntington disease. In a study of a genetically homogeneous group of 41 subjects with the 460InsA mutation in FTL1, the mean age of onset was 39.4 years, beginning with chorea in 50%, focal lower limb dystonia in 42.5% and parkinsonism in 7.5% of the patients [70]. The majority reported a family history of a movement disorder often misdiagnosed as Huntington disease. The disease progressed relentlessly, becoming generalized over a 5- to 10-year period, eventually leading to aphonia, dysphagia and severe motor disability with subcortical/frontal cognitive dysfunction as a late feature. A characteristic action-specific facial dystonia was common (65%), and in 63% of patients there was asymmetry throughout the disease course. Serum ferritin levels were low in the majority of males and postmenopausal females, but within normal limits for premenopausal females. Isolated parkinsonism is unusual in neuroferritinopathy, and unlike Huntington’s disease, cognitive changes are absent or subtle in the early stages. Depressed serum ferritin is common and provides a useful screening test in routine practice [70].
Dentatorubropallidoluysian atrophy is an autosomal dominant disorder characterized by chorea (choreoathetosis), ataxia, dementia, seizures, myoclonus, and dystonia [53,191,357]. The disorder is particularly prevalent in Japan. A possibly similar disorder has been described in the southern United States as the “Haw River” syndrome [56]. The usual age of onset is the fourth decade (range, first to seventh decade). An early onset subtype presents with a variable combination of myoclonus, epilepsy, and mental retardation, whereas a late-onset subtype is characterized by choreoathetosis, cerebellar ataxia, dystonia, rest and postural tremor, parkinsonism, and dementia. Pathologically patients demonstrate degeneration affecting the dentatorubral system, globus pallidus, subthalamus, and, to a lesser extent, the striatum, substantia nigra, inferior olive, and the thalamus.
Familial dyskinesia and facial myokymia (FDFM) is a disorder that has an early childhood or adolescent onset and is characterized by adventitious movements that sometimes appear choreiform and that are associated with perioral and periorbital myokymia [117]. The involuntary movements are paroxysmal at early age, increase in frequency and severity, and may become constant by the third decade. Thereafter, there is no further deterioration and there may even be improvement in old age. Although this familial entity may be confused with Huntington’s disease, there is no intellectual impairment and the lifespan is normal.
Sydenham chorea (rheumatic chorea or St Vitus dance), related to group A beta-hemolytic streptococcal infection, occurs in childhood and adolescence. Sydenham chorea is predominantly or exclusively unilateral (hemichorea) in about half of the cases, and it may result in an apparent flaccid paralysis (chorea mollis). HIV encephalitis may be a cause of chorea [130,271] as may tuberculous meningitis and herpes simplex encephalitis [239]. Chorea may also be caused by medications (e.g., l-dopa, neuroleptics, metoclopramide, anticonvulsants, propofol, pemoline, corticosteroids, lithium, digoxin) and drugs of abuse (e.g., cocaine-induced choreoathetosis or “crack dancing” [86]; amphetamines). Chorea may be associated with a variety of systemic disorders (e.g., systemic lupus erythematosus, Henoch-Schönlein purpura, sarcoidosis, vasculitis, multiple sclerosis, Behçet disease, hyperthyroidism, Hashimoto encephalopathy, hypoparathyroidism, acquired hepatocerebral degeneration, renal failure, polycythemia vera, hypo- or hypercalcemia, hypo- or hypernatremia, hypo- or hyperglycemia, mercury poisoning, carbon monoxide poisoning), and may also occur in association with the primary antiphospholipid antibody syndrome. Chorea with dementia may occur from paraneoplastic striatal encephalitis [339] and paraneoplastic chorea may be associated with CRMP-5 neuronal antibody and lung carcinoma [348]. Chorea may also be seen in variant Creutzfeldt-Jakob disease (vCJD). A physiologic form of chorea is present in some infants. Chorea may occur in children as a sequelae of cardiac surgery (“postpump chorea”), especially associated with prolonged time on the pump, deep hypothermia, or circulatory arrest [256], and choreoathetosis and orofacial dyskinesia, hypotonia, and pseudobulbar palsy (CHAP syndrome). CHAP syndrome may occur after surgery for congenital heart disease [137]. A steroid-responsive chorea has been described after heart transplant [43]. Chorea may also be seen in pregnancy (chorea gravidarum), and with the administration of oral contraceptives [246], congophilic angiopathy may cause chorea associated with progressive dementia [36]. Chorea is the commonest movement disorder following a stroke and appears in older patients [5]. After a stroke, involuntary movements tend to persist despite the functional recovery of motor deficit.
The pathogenesis of choreiform movements is essentially unknown. The evidence linking these abnormal movements to the caudate and putamen is by no means convincing, because most associated disease processes (e.g., Huntington’s disease) show diffuse or multiple lesions that affect other neural structures. Dystonia and choreoathetosis are rare associations of cervical cord lesions, such as ependymoma, glioma, myxoma, demyelination, trauma, and cervical disc prolapse [338].
Unilateral chorea (hemichorea) is customarily seen with lesions of the contralateral subthalamic nucleus of Luys or its connections, although it has also been known to occur with lesions of the thalamus or caudate nucleus. The choreic movements may involve the entire half of the body or may spare the face. It is often fruitless and impractical to distinguish hemichorea from hemiballismus; in fact, the two disorders probably represent opposite ends of a spectrum of hyperkinesias. Hemichorea may be seen with infarction or hemorrhage. It may also occur as a complication of thalamotomy or, rarely, secondary to moyamoya, trauma, migraine, or neoplasm. Hemichorea on the right side has been described with a left putaminal cavernous angioma [104].
An overview of the numerous causes of chorea is outlined in Table 19.1.
Tardive Dyskinesia and Other Tardive Syndromes
Tardive dyskinesia and other tardive syndromes result from treatment with neuroleptic drugs and other dopamine receptor blocking agents, including drugs used for gastrointestinal problems (metoclopramide), depression (amoxapine, perphenazine/ amitriptyline), and cough (promethazine). Onset after exposure of less than 3 months is possible but uncommon [83]. The abnormal involuntary movements can appear when the patient is taking the drug or after stopping the drug and may persist and can even remain permanently. Withdrawing the offending drug often exacerbates the severity of the movements, whereas increasing the dosage often ameliorates the movements.
A variety of movement abnormalities may occur as tardive syndromes. The most common tardive dyskinesia pattern is repetitive, almost rhythmical movements that can be labeled stereotypic and most often occurs in the oral-lingual-buccal area. These oral-buccal-lingual dyskinesias usually present as complex chewing movements often associated with occasional popping out of the tongue and with writhing movements of the tongue at rest in the mouth [165]. Other parts of the body, such as the hands, feet, and trunk, may also develop rhythmical movements and the abdominal and pelvic muscles may be affected, resulting in truncal or pelvic rocking or thrusting movements. Respiratory dyskinesias can result in involuntary chest and diaphragmatic movements [112]. Tardive dyskinesia is often accompanied by a feeling of unpleasant inner restlessness (akathisia) that can be whole body restlessness or uncomfortable sensations in a specific part of the body [40,55]. The third most common movement pattern is tardive dystonia, which may be accompanied by tardive akathisia or tardive dyskinesia [54,55,183,345]. With tardive dystonia, the abnormal movements are more sustained and sometimes torsional in nature. Facial dystonia may occur in the form of blepharospasm or facial grimacing, whereas involvement of the mandible can result in tonic jaw deviation, jaw protrusion, sustained opening and closing of the jaw, or bruxism. Dystonic posturing of the neck is also common, particularly retrocollis [178]. Tardive dystonia can occur at all ages, whereas classic tardive dyskinesia is more common in the elderly. Other abnormal movements that may occur less commonly as tardive syndromes include myoclonus, tremor, oculogyric crisis, and tics.
TABLE 19.1 Causes of Chorea

Orofacial Dyskinesia
Orofacial dyskinesias [7] are abnormal involuntary movements of the facial musculature, lips, and tongue that may appear spontaneously, especially in elderly edentulous patients, or in Huntington disease, Sydenham chorea, or Wilson disease. Their occurrence after prolonged neuroleptic therapy (tardive dyskinesia) [165] favors an etiology involving denervation supersensitivity of the striatum. A unilateral striatonigral lesion may produce bilateral orofacial-lingual dyskinesia plus contralateral hemidystonia, suggesting that the basal ganglia of one hemisphere may exert bilateral orofacial-lingual motor control [164]. The differential diagnosis of orofacial dyskinesia is outlined in Table 19.2.
TABLE 19.2 Differential Diagnosis of Orofacial Dyskinesia

Abdominal Dyskinesias
Abdominal dyskinesias are continuous movements of the abdominal wall or sometimes the diaphragm [161,195]. Their sinuous, rhythmic nature has led to them being called belly dancer’s dyskinesia. Abdominal dyskinesia may occur after abdominal trauma (e.g., laparotomy) in some cases and may be associated with abdominal myoclonus. These dyskinesias may also occur as a tardive syndrome.
Ballismus
Ballismus [185,230,351] is an involuntary hyperkinesia often confined to one half of the body (hemiballismus), but it may involve a single extremity (monoballismus) or, exceptionally, both halves of the body (paraballismus or biballismus). Hemiballismus is characterized by the occurrence of sudden, paroxysmal, large-amplitude, flinging, throwing movements of the arm and leg contralateral to a lesion in or near the subthalamic nucleus. These abnormal movements are often continuous during wakefulness and cease with sleep. Hemiballismus is often associated with decreased muscle tone in the involved extremities.
Hemiballismus usually occurs with lesions that affect the contralateral subthalamic nucleus of Luys [230] or disrupt the afferent or efferent connections of this structure. Transient hemiballismus/hemichorea has been described with an ischemic lesion of the ipsilateral subthalamic nucleus [82]. Hemiballismus may also result from lesions in the caudate, putamen, globus pallidus, precentral gyrus, or thalamic nuclei [201,260]. The simultaneous occurrence of hemiballismus with acute ipsilateral central pain has been described after anterior parietal artery stroke [304]. Hemiballismus/hemichorea has been described in patients with nonketotic hyperglycemia with magnetic resonance imaging (MRI) studies revealing high signal intensity in the contralateral striatum [209]. Hemiballismus is most commonly caused by a discrete ischemic or hemorrhagic vascular lesion of the subthalamus [351]. It is uncertain whether the occluded vessels arise from the posterior thalamoperforating, posterior communicating, or anterior choroidal arteries [120]. Hemiballismus has also been noted with tumors, arteriovenous malformations, encephalitis, abscess, systemic lupus erythematosus, acquired immunodeficiency syndrome (AIDS), cysticercosis, head trauma, subdural hematoma, tuberculous meningitis, demyelination, tuberous sclerosis, Sydenham chorea, nonketotic hyperglycemia, basal ganglia calcifications, multiple systems atrophy (MSA), as a side effect of levodopa therapy, oral contraceptives, or after surgery for advanced Parkinson disease (PD) [67,102,140,260,295,351]. Bilateral ballismus has been described after bilateral basal ganglia hemorrhagic infarcts involving the caudate and putamen, with multiple sclerosis, with disseminated intravascular cancer, with systemic lupus erythematosus, after ventriculoperitoneal shunting, with nonketotic hyperglycemia, with phenytoin intoxication, and with dopaminergic drug-induced dyskinesia in PD [222,287].
Akathisia
Akathisia refers to a feeling of inner restlessness that is often relieved by movement [40,134]. The motor activity is, therefore, described by patients as a voluntary effort to relieve uncomfortable sensations, although in severe cases the need for motor activity is beyond control. Akathitic movements are typically complex and stereotyped with movements including “squirming” in a chair, repetitive shifting of weight, crossing and uncrossing the legs, inability to remain seated, pacing, rocking the trunk, and even moaning, humming, or groaning. Other movement disorders associated with moaning sounds or humming include tics, oromandibular dystonia, Huntington’s disease, and parkinsonism [134,262]. A specific body part may be affected, with relief of the perceived discomfort (burning or pain) attainable by movement. Common sites for the discomfort are the mouth and vagina [121].
Akathisia is most often due to medications, especially agents that block dopamine receptors (e.g., neuroleptics, antiemetics, tetrabenazine, reserpine). It can occur with initiation of drug treatment (acute akathisia), subsequently with the emergence of drug-induced parkinsonism, or after chronic treatment (tardive akathisia), with the latter usually made worse by drug withdrawal. Akathisia may also be present in patients with parkinsonism.
A distinct clinical syndrome referred to as hypotensive akathisia has been described in patients with autonomic failure who manifest habitual, voluntary, transiently suppressible, yet irresistible leg movements occurring only in the sitting position [69]. Repetitive leg crossing, muscle tensing, foot twirling or wiggling, or heel or toe tapping while sitting may have compensated for orthostatic hypotension and raised systolic and diastolic blood pressure.
Athetosis
Athetosis is characterized by slow, uncoordinated, twisting, writhing, involuntary movements of wide amplitude. These movements predominantly involve the distal appendicular musculature, especially the upper extremities, although facial and axial muscles may also be involved.
Athetoid movements may be unilateral (hemiathetosis) or bilateral (double athetosis) and may interfere considerably with activities of daily living. These movements are often associated with episodic muscular hypertonia affecting the axial and appendicular muscles. The differentiation of athetosis from chorea and dystonia may at times be difficult because it is not uncommon to see a patient with “mixed” dyskinesias (i.e., choreoathetosis).
Athetosis is usually noted with degenerative disorders (e.g., Wilson’s disease, kernicterus, status marmoratus, perinatal anoxia) involving widespread cerebral structures, including the globus pallidus, subthalamus, red nucleus, and midbrain tegmentum. A focal lesion (vascular or neoplasm) that damages the striatum but spares the motor cortex and its efferents may rarely cause athetosis.
Athetoid movements must be differentiated from “pseudoathetoid” movements, which are noted on attempts to maintain posture (e.g., extending the arms) and are due to faulty proprioception (as in lesions affecting the large peripheral nerve fibers, the dorsal root ganglia, the posterior columns and their connections, or the parietal lobe) [324].
Dystonia
Dystonia is characterized by slow, long-sustained, contorting, involuntary movements and postures involving proximal appendicular and axial muscles. The dystonic movements are typically slow and “wrapping” (athetotic dystonia), although patients may sporadically demonstrate superimposed, rapid, involuntary jerks termed dystonic spasms or myoclonic dystonia. The dystonic posturing results in disabling and abnormal attitudes (dystonic postures) of the affected body parts (e.g., torticollis, tortipelvis, lordotic or scoliotic postures, inversion of the hands and forearms, equinovarus deformity) [196]. Therefore, the characteristic features of dystonia include: (a) excessive co-contraction of antagonist muscles during voluntary movement, (b) overflow of contraction to remote muscles not normally employed in voluntary movement, and (c) spontaneous spasms of co-contraction [241]. Dystonia can also be associated with fast, rhythmic, tremulous movements (dystonic tremor) [173,302]. Dystonic tremor of the hand has been described after contralateral anterior thalamic infarction [71]. Oculogyric crisis, thought to represent dystonia of the ocular muscles, has been described with the use of neuroleptics, neuroleptic malignant syndrome, Wilson disease, encephalitis lethargica, organophosphate poisoning, and with lesions of the lentiform nuclei [179].
Dystonia may be generalized and idiopathic (dystonia musculorum deformans) or symptomatic (drug-induced, Wilson disease, Menkes disease, metazchromatic leukodystrophy, Lesch-Nyhan disease, homocystinuria, hexosaminidase A and B deficiencies, progressive supranuclear palsy (PSP), cortical-basal ganglionic degeneration (CBGD), GM1 and GM2 gangliosidosis), or it may be segmental, affecting only one body part, and idiopathic (spasmodic torticollis, writer’s cramp, musician’s cramp or dystonia, spasmodic dysphonia (SD), blepharospasm, orofacial dyskinesia) or symptomatic (posthemiplegic dystonia). Occasionally, dystonia may be focal and task specific (i.e., brought on only by specific activities or occupations, such as writing, typing, playing golf, playing the piano, blowing a horn [236], etc.), and associated with a task-specific tremor [302]. One unusual form of occupational-specific oromandibular hemidystonia is “auctioner’s jaw” [320]. Patients with focal task-specific dystonia demonstrate cortical sensory representation that is markedly abnormal and inappropriately activate cortical regions (typically the supplementary motor cortex) that are not activated during normal performance of manual tasks. Acute dystonic camptocormia (bending of the trunk) has been described with lenticular, mostly putaminal, vascular lesions [275].
Lesch–Nyhan disease is caused by deficiency of the purine salvage enzyme hypoxanthine-guanine phosphoribosyltransferase (HPRT) [175]. Affected individuals exhibit overproduction of uric acid, along with a characteristic neurobehavioral syndrome that includes mental retardation, recurrent self-injurious behaviour and motor disability. In one review of this syndrome, 44 patients ranging from 2 to 38 years presented a characteristic motor syndrome that involved severe action dystonia superimposed on baseline hypotonia [175]. Although some patients also displayed other extrapyramidal or pyramidal signs, these were always less prominent than dystonia.
Hemidystonia may follow lesions of the contralateral caudate, lentiform nucleus (especially the putamen), or thalamus, or a combination of these structures [75,128,245,269,371]. It may be due to abnormal input from the thalamus to the premotor cortex due to lesions either of the thalamus itself or of the striatum projecting by way of the globus pallidus to the thalamus [245]. The most common etiologies of hemidystonia include stroke, trauma, and perinatal injury [75]. In one study, lesions associated with dystonic spasms or myoclonic dystonia tended to be located in the striatopallidal complex or thalamus contralateral to the dystonia [213]. Lesions of the striatopallidal complex involved the putamen posterior to the anterior commissure and extended variably into the dorsolateral caudate nucleus, the posterior limb of the internal capsule, or the lateral segment of the globus pallidus. These lesions were centered in the “sensorimotor” part of the striatopallidal complex with a trend toward a somatotopical distribution. Lesions of the thalamus were located in the ventral intermediate and ventral caudal nuclei, whereas the ventral oral anterior and posterior nuclei (which receive pallidal efferents) are largely spared [213]. Paroxysmal hemidystonia may occur with contralateral midbrain lesions [322].
Beside focal lesions of the basal ganglia and thalamus, brainstem lesions may also be associated with dystonia [223]. Four patients were reported with acquired dystonia following brainstem lesions [223]. Three patients suffered tegmental pontomesencephalic hemorrhage and one patient diffuse axonal injury secondary to severe craniocerebral trauma. Dystonia developed with a delay of 1 to 14 months, at a mean delay of 6 months. All patients presented with hemidystonia combined with cervical dystonia, and two patients had craniofacial dystonia in addition. Three patients had postural or kinetic tremors. Dystonia was persistent in three patients, and improved gradually in one. Overall, the phenomenology of secondary dystonia due to pontomesencephalic lesions is similar to that caused by basal ganglia or thalamic lesions. Structures involved include the pontomesencephalic tegmentum and the superior cerebellar peduncles [223].
Hemidystonia-hemiatrophy syndrome is a very disabling neurological condition similar to the hemiparkinsonism-hemiatrophy syndrome [365]. In a study of 26 patients (14 female) with a mean age at onset of hemidystonia at 14.9 years (1–46 years) the mean latency from the onset of hemiparesis to the onset of hemidystonia was 14.7 years (2 weeks– 46 years) [365]. All patients with hemiparesis had marked improvement in their weakness prior to the onset of hemidystonia. Common causes leading to hemiparesis and subsequent hemidystonia were birth or perinatal complications (N = 13) and stroke (N = 10). Hemidystonia-hemiatrophy is usually associated with static encephalopathy originating at very young age, but the syndrome may also represent delayed sequelae of a stroke or brain injury [365].
Primary dystonias may be of genetic origin. Hereditary childhood-onset dystonia (idiopathic torsion dystonia, DYT1) most commonly starts between age 6 and 12 years with dystonia of the foot while walking. The illness is slowly progressive and the dystonia becomes generalized. The disorder is usually autosomal dominant with reduced penetrance. The abnormal gene, located on chromosome 9q, produces a protein called torsin A with unknown function [283]. Other autosomal dominant dystonias include DYT4, DYT6, DYT7, and DYT13 [90]. DOPA-responsive dystonia (Segawa variant or hereditary progressive dystonia) is an autosomal dominant disorder linked to chromosome 14q22.1-q22.2. This disorder typically presents in childhood (age 5–6 years) with dystonic movements and postures that are remarkably responsive to low doses of levodopa [152,321]. These children may also have features of parkinsonism, lower extremity hyperreflexia, and bilateral extensor plantar responses often misdiagnosed as “cerebral palsy.” The dystonic movements and postures may show marked diurnal variations, being more pronounced in the late afternoon, evening, and night. The etiology for the disorder is most commonly a mutation in the GTP cyclohydrolase I gene, which leads to a deficiency in the production of dopamine [160]. Since the gene has been identified, the clinical spectrum of the disorder has been expanded to include adult-onset parkinsonism, oromandibular dystonia, spontaneously remitting dystonia, spasticity with developmental delay mimicking cerebral palsy, and generalized hypotonia with proximal weakness [90]. Patients with autosomal dominant Segawa syndrome due to GTP cyclohydrolase deficiency also may manifest late-presenting mild dopa-responsive symptoms of rigidity, frequent falls, and tendonitis [346]. Major depressive disorders, often recurrent, obsessive-compulsive disorder, and sleep disorders, including difficulty in sleep onset and maintenance, excessive sleepiness, and frequent nightmares, may also occur in these patients [346]. Cerebellar signs and scoliosis has also been described in some patients with dopa-responsive dystonia [64].
DYT14 is a genetic defect that can also lead to dopa-responsive dystonia [146]. Myoclonic dystonia (DYT11) is an autosomal dominant syndrome where symptoms include dystonic myoclonus as well as more prolonged spasms [132,182]. The clinical manifestations generally occur in the first or second decade of life. In this setting, myoclonus is usually the main and most disabling feature; it predominates in the arms and axial muscles and is often alcohol-responsive. Dystonia is usually mild and often manifests as cervical dystonia or writer’s cramp. Tremor, similar to essential tremor or action tremor, may also be present. There is often a marked response to ethanol. In many families, the genetic abnormality has been identified to be in the protein epsilon-sarcoglycan [20,299]. There is an X-linked recessive dystonia-parkinsonism called Lubag primarily found in the Philippine Islands [359]. Rapid-onset dystonia parkinsonism (DYT12) is a striking autosomal dominant disorder characterized by abrupt onset of dystonia, usually accompanied by signs of parkinsonism. The sudden onset of symptoms over hours to a few weeks, often associated with physical or emotional stress, suggests a trigger initiating a nervous insult that results in permanent neurologic disability [91]. Autosomal dominant dystonia-plus with cerebral calcifications may present with focal, segmental, multifocal, or generalized dystonia sometimes associated with chorea, intellectual decline, postural tremor, and dysarthria [368].
Woodhouse Sakati syndrome is a rare autosomal recessive neuroendocrine disorder characterized by the combination of alopecia, hypogonadism, diabetes mellitus, mental retardation, sensory neural deafness and extrapyramidal features [314]. Movement disorders mainly consist of dystonia and chorea of the limbs with onset in adolescence [314]. Facial muscles are usually spared, but dysarthria is common. Pyramidal features and peripheral abnormalities are inconsistent features. Most of the reported families are from the Middle Eastern countries although rarely Caucasian cases have been described.
An unusual familial dystonia-plus phenotype characterized by dystonia and cerebellar atrophy on brain MRI has been described in 12 patients in eight families [208]. The mean age at onset was 27.3 ± 11.5 years (range: 9–42 years). At onset, dystonia was focal or multifocal, mainly affecting vocal cords and upper limbs. During the disease course spasmodic dysphonia became severe in five patients, leading to complete aphonia in two. Dystonia became generalized in five patients. Cerebellar ataxia was limited to unsteadiness in most patients and progressed very slowly. The paucity of clinical cerebellar signs contrasted with the marked cerebellar atrophy on brain MRI in most patients. Four families with two affected sibs support the hypothesis of an autosomal recessive disorder. However, X-linked inheritance is possible because only men were affected [208].
Focal dystonias (e.g., torticollis) are usually sporadic and occur in later life but patients may have more than one form of dystonia. There may well be a genetic basis for focal dystonias; for example, one family with spasmodic torticollis (DYT7) had a genetic linkage to chromosome 18p [215]. Secondary dystonias can be caused by a variety of neurologic disorders including Parkinson disease, Wilson disease, gangliosidoses, leukodystrophies, Leigh syndrome, Hallervorden-Spatz disease, the juvenile form of Huntington disease, corticobasal ganglionic degeneration, and brain lesions affecting the putamen, caudate, and thalamus. Dystonia can also be psychogenic [207].
Schrag et al. described the clinical features of 103 patients presenting with “fixed dystonia” [318]. Most patients were female (84%) and had a young age of onset (mean 29.7 years). A peripheral injury preceded onset in 63% and spread of dystonia to other body regions occurred in 56%. After an average follow-up of 3.3 years (overall disease duration 8.6 years), partial (19%) or complete (8%) remission had occurred in a minority of patients. The fixed postures affected predominantly the limbs (90%), and rarely the neck/shoulder region (6%) or jaw. The authors conclude that fixed dystonia often occurs after a peripheral injury and that whether the disorder is primarily neurologic or psychiatric remains an open question.
A classification of dystonias is outlined in Table 19.3.
Torticollis
Torticollis (idiopathic cervical dystonia, wryneck, nuchal dystonia) [11,45,87,170,204,229] is a hyperkinesia slightly more frequent in women and characterized by tonic or clonic contraction of the neck musculature, especially the sternocleidomastoid and trapezius muscles. This type of cervical dystonia results in a more or less stereotyped fixed or spasmodic deviation of the head into an anomalous position with the chin twisted to one side or the head displaced backward (retrocollis) or forward (anterocollis) in about a quarter of the patients, laterally (laterocollis) in half of them, or, most often, in a combination of these abnormal postures. By convention, spasmodic torticollis is named by the sternocleidomastoid muscle that contracts. Spasmodic torticollis is usually unilateral, ceases during sleep, and increases with anxiety and stress. Often, the abnormal movement can be relieved by sensory tricks, such as a light touch on the face (the geste antagoniste). Other effective maneuvers include leaning against a high-backed chair, placing something into the mouth, or pulling the hair [87]. Local pain is reported by most patients. About a third of them have scoliosis or a secondary cervical radiculopathy. In addition to cervical dystonia, approximately 10% to 20% of these patients have oral, mandibular, or hand-arm dystonia or blepharospasm [87,170]. Tremor, particularly involving the head, is present in approximately 60% of the patients [170].
TABLE 19.3 Classification of Dystonias

Torticollis may be congenital, tardive, secondary to acquired cervical spine abnormalities (e.g., cervical spondylosis, subluxation of the cervical spine, inflammatory disorders of the cervical spine), or due to upper spinal cord tumor with syringomyelia [184]. Posttraumatic cervical dystonia may develop immediately after relatively mild trauma to the neck [142]. Cervical dystonia may also rarely occur as a sign of a posterior fossa tumor and retrocollis has been described with bilateral putaminal hemorrhages [326]. Focal dystonia may also occur with lesions of the lenticular and caudate nuclei, especially infarcts and tumors [199]. Familial dopa-responsive cervical dystonia has been described and a trial of levodopa should be considered in patients with young-onset cervical dystonia [316]. Transient cervical dystonia may rarely occur during pregnancy (dystonia gravidarum) [76,218]. Cervical dystonia occurs, however, most often as an extrapyramidal disorder of unknown etiology and pathologic substrate (spasmodic torticollis or idiopathic cervical dystonia) and is the most common form of adult-onset focal dystonia [87].
WRITER’S CRAMP, MUSICIAN’S DYSTONIA, THE YIPS, AND OTHER FOCAL DYSTONIAS
Writer’s cramp (graphospasm) [174,177,325] is a segmental dystonia characterized by spasms, cramps, aches, and occasional tremors of the hand muscles induced by writing. Examination reveals no evidence of oromandibular, axial, or appendicular dystonia, blepharospasm, or torticollis. Musician’s dystonia is a focal task-specific dystonia induced only by playing certain musical instruments [166,303,319]. Focal hand dystonia has been suggested to be a maladaptive response of the brain to repetitive performance of stereotyped and attention-demanding hand movements [303]. However, not all patients with focal hand dystonia have a strict history of excessive hand use; for example, patients with musician’s dystonia spend many hours per day with their attention focused on instrumental practice, whereas many patients with writer’s cramp have a history of average hand use. Mirror dystonia (dystonia occurring in the dominant hand when writing with the other hand) was noted in 29 of 65 patients with writer’s cramp in one study [174]. The etiology and pathologic substrate are unknown. Some authors have suggested that this dystonia may be due to an abnormality of the contralateral primary and secondary supplementary motor areas resulting in dysfunction in motor programming [100]. Structural abnormalities in brain structures interconnected within the sensorimotor network, including the cerebellum and the cortical representation of the affected hand, have been demonstrated in patients with writer’s cramp [94]. These abnormalities may be related to the pathophysiology of writer’s cramp, questioning the role of the cerebellum, or maladaptive plasticity in a task-related dystonia [94].
In a case-control study of risk factors for writer’s cramp in 104 consecutive patients, cases had a college or university degree more frequently than controls [307]. The risk of writer’s cramp increased with the time spent writing each day and was also associated with an abrupt increase in the writing time during the year before onset. Head trauma with loss of consciousness and myopia were both associated with the condition but it was not significantly associated with peripheral trauma, left-handedness, constrained writing, writing in stressful situations or the choice of writing tool. The dose–effect relationship between writer’s cramp and the time spent handwriting each day, and the additional burden of acute triggers such as an abrupt increase in the writing time in the year before onset, suggest that writer’s cramp in a disruptive phenomenon in predisposed subjects [307].
Up to 30% of golfers develop the yips, an inability to complete a golf stroke, most often affecting short putts, which worsens with anxiety [2,251]. Yips may be organic (a task-specific dystonia) or psychological (anxiety or “choking”). The finding that yips-affected golfers often have co-contraction of wrist flexors and extensors suggests that in many individuals this disorder is a task-specific dystonia [2]. Table tennis dystonia is another form of task-specific dystonia [212].
Patients have been described who presented with the acute onset of a movement disorder characterized by a tonic, sustained, lateral and outward protrusion of one half of the lower lip [188]. The movement disorder was present at rest, whereas in some patients, it was also present during speech. In all cases, the abnormal lip posture could be suppressed voluntarily and spontaneous remissions were frequent. This may well represent a focal lip dystonia.
Intermittent or sustained severe involuntary tongue protrusion dystonia may cause speech, swallowing, and breathing difficulties that can be severe enough to be life threatening [313]. Causes include neuroacanthocytosis, pantothenate kinase-associated neurodegeneration, Lesch-Nyhan syndrome, and postanoxic and tardive dystonia. Oromandibular dystonia may involve the lateral pterygoid muscles causing incapacitating protrusive and lateral jaw movements and displacements [265].
Frucht reported 89 musicians with focal task-specific dystonia of the embouchure (ED), the muscles of the lower face, jaw, and tongue used to control the flow of air into the mouthpiece of a woodwind or brass instrument [129]. Symptoms of ED began at an average age of 36, were typically painless, and only rarely were preceded by trauma. Specific musical techniques commonly triggered dystonia, often in one instrumental register. Task-specific embouchure tremor and lip-pulling ED phenotypes were common among high-register brass players (trumpet and French horn), whereas lip-locking occurred exclusively in low-register brass players (trombone and tuba). Jaw and tongue ED phenotypes occurred predominantly in woodwind players, and once present, frequently spread to speaking or eating. Six percent of all ED patients had coincident writer’s cramp, suggesting a possible genetic predisposition to develop dystonia. Once present, symptoms of ED did not remit and often disrupted careers and livelihoods [129].
A middle cerebral artery distribution stroke may cause contralateral hand weakness and gradually severe dystonia emerging in the hand with spreading of fingers in a “starfish” pattern. (“starfish hand”), likely due to caudate head involvement by the stroke [157].
The lower extremity is affected infrequently in adult-onset primary dystonia in contrast to childhood-onset dystonia, which typically begins in the foot. When dystonia affects the foot in an adult, it is usually on a secondary basis. Schneider et al. reviewed the findings on 17 patients (11 women, 6 men; average age of onset 48.4 years; average time to diagnosis 2.7 years) with adult-onset primary foot dystonia [315]. The most common patterns were plantar flexion of all toes and inversion of the foot, typically activated with standing or walking.
Another form of adult-onset focal dystonia occurs in athletes (runner’s dystonia) and is often wrongly attributed to an orthopedic disorder [369]. These patients develop proximal dystonia of one leg during long-distance running. The clinical features of dystonia in these long-distance runners overlap with those of more recognizable forms of focal dystonia including relief with sensory or motor “tricks”. These patients differed from the typical childhood-onset leg dystonia, such as the DYT1 dystonia, in that there is no family history of dystonia and the leg dystonia remains focal without spreading to other body parts [369].
BLEPHAROSPASM
Blepharospasm is characterized by intermittent or sustained spontaneous forceful eye closure that may render the patient functionally blind [60,168]. These movements may occur in patients with parkinsonism or torsion dystonia or as a side effect of neuroleptic drugs. Blepharospasm has been described with dorsomedial pontine tegmental or upper brainstem lesions [12,199]. Blepharospasm is also seen with oromandibular dystonia, as in Meige syndrome (idiopathic blepharospasm-oromandibular dystonia) [17,168,240,344], a condition probably related to dopaminergic predominance in the striatum.
SPASMODIC DYSPHONIA
Spasmodic dysphonia (SD) [10,119,250,289] or laryngeal dystonia is a disorder of unknown etiology characterized by a tremulous, forced voice with a low tone and volume and often associated with facial grimacing. Three subtypes of SD are described by perceptual and acoustic vocal characteristics: adductor, abductor, and mixed [13–16,58]. Adductor SD is associated with involuntary hyperadduction of the vocal folds, resulting in a strained or strangled voice quality. Abductor SD is characterized by involuntary abductions of the vocal folds, resulting in intermittent bursts of breathy phonation. Mixed SD is applied to patients who exhibit a full range of these vocal characteristics. Pitch breaks, hoarseness, limited intensity range, and poor intensity control are present in all three types [289]. It has been proposed that SD is a continuum disorder in which both types of spasms occur with differing frequencies [58].
Patients with SD often have abnormal neurologic examinations, including abnormal rapid alternating movements and tremor, including voice tremor [14–16,289]. Some authors view SD as an incomplete expression of Meige syndrome [168], as an adult-onset focal dystonia [232], or as an abnormality of the motor control system that includes the globus pallidus, putamen, thalamus, and supplementary motor area [119,210,250,289].
Primary craniocervical dystonia may present as a respiratory emergency [286]. A similar phenomenon has been reported in a subtype of isolated laryngeal dystonia named spasmodic laryngeal dyspnea [373]. This disorder differs from spasmodic dysphonia in that symptoms are dependent on respiration rather than phonation. In these cases, dyspnea is caused by an intermittent glottic and supraglottic airway obstruction from both laryngeal and supralaryngeal/pharyngeal muscle spasms. Acute laryngeal dystonia has been identified as a life-threatening side effect of classic antipsychotics [74].
Paroxysmal Dyskinesias
Paroxysmal dyskinesias are a heterogeneous group of disorders that have in common sudden abnormal involuntary movements out of a background of normal motor behavior. The abnormal movements may be choreic, ballistic, dystonic, or a combination of these. They consist of episodic attacks of involuntary movements, may be classified according to phenomenology, duration of attacks, and etiology [34,51,95,113,176,205], and include the following.
1. Paroxysmal kinesigenic dyskinesia (PKD) (paroxysmal kinesigenic choreoathetosis). In these patients, attacks of abnormal involuntary movements (typically lasting seconds to minutes) occur abruptly after a sudden voluntary movement or startle. Hyperventilation may also induce an episode. Ballism, dystonic postures, chorea, athetosis, or any combination of these make up the movements, which are occasionally preceded by paresthesias, tenseness, or crawling sensations. The attacks may cause falls and may affect speech. The abnormal movements are easily habituated and therefore fail to recur if the sudden movement is immediately repeated. Although this abnormality is usually idiopathic [51], it may occur on a hereditary basis and has also been described with multiple sclerosis, head trauma, PSP, putaminal/ thalamic infarction, hypoparathyroidism with basal ganglia calcifications, HIV infection, and hyperglycemia with lenticular vascular malformation [41,264,300]. Families with members that have PKD may have other members with infantile convulsions, suggesting a shared PKD/infantile convulsions gene [337]. Approximately 90% of patients improve with medications, especially anticonvulsants. Bruno et al. reviewed the clinical features of 121 affected individuals with a presumptive diagnosis of idiopathic PKD [51]. The majority (79%) of affected subjects had a distinctive homogeneous phenotype. The authors propose the following diagnostic criteria for idiopathic PKD based on this phenotype: identified trigger for the attacks (sudden movements), short duration of attacks (<1 minute), lack of loss of consciousness or pain during attacks, antiepileptic drug responsiveness, exclusion of other organic diseases, and age at onset between 1 and 20 years if there is no family history (age at onset may be applied less stringently in those with family history). In comparing familial and sporadic cases, sporadic cases were more frequently male, and infantile convulsions were more common in the familial kindreds. Females had a higher remission rate than males. An infantile-onset group with a different set of characteristics was identified. A clear kinesigenic trigger was not elicited in all cases, antiepileptic response was not universal, and some infants had attacks while asleep.
2. Paroxysmal nonkinesigenic dyskinesia (paroxysmal dystonic choreoathetosis). With this entity, attacks of involuntary movements (usually lasting minutes to hours) occur spontaneously and consist of combinations of dystonic posturing, chorea, athetosis, and ballism [172]. Speech is often affected and attacks may be preceded by paresthesias, stiffness, or crawling sensations. These attacks may occasionally be triggered by stress, fatigue, excitement, caffeine, or alcohol. Although usually idiopathic, this entity often occurs on a hereditary basis and has also been described with multiple sclerosis, perinatal encephalopathy, hypoparathyroidism with basal ganglia calcification, encephalitis, thyrotoxicosis, stroke, infantile hemiplegia, head trauma, hypoglycemia, AIDS, diabetes mellitus, anoxia, and brain tumor [41,264]. For example, paroxysmal nonkinesigenic dyskinesias have been described because of recurrent hypoglycemia caused by an insulinoma [89]. This abnormality may also occur on a functional basis. Not sensitive to anticonvulsants, only a third of the patients improve with medications.
3. Paroxysmal exertion-induced dyskinesia. With this form, attacks are precipitated by prolonged physical exertion. The abnormal movements especially affect the legs.
All three types may be further subdivided into short-lasting (≤5 minutes) and long-lasting (>5 minutes) attacks.
4. Paroxysmal hypnogenic dyskinesia. In this form, episodes of involuntary movements occur only during sleep.
5. Infantile convulsions and choreoathetosis syndrome (ICCA syndrome). Families with this syndrome have members that suffer infantile convulsions and later develop episodes of paroxysmal choreoathetosis [34]. Attacks of choreoathetosis resemble PKD in being very brief and frequent and induced by sudden exertion.
Many of the hereditary forms of paroxysmal dyskinesia may be due to channelopathies [34,35]. Other paroxysmal movement disorders that have been described include benign paroxysmal dystonia/torticollis in infancy (Sandifer’s syndrome) and paroxysmal ataxia and tremor. Sandifer’s syndrome is characterized by spasmodic posturing of the head and neck as a result of gastroesophageal reflux [235]. Paroxysmal ataxia and tremor may be associated with persistent limb myokymia or neuromyotonia, nystagmus, or ocular motility dysfunction.
Myoclonus
Myoclonus [3,32,61,85,171,243,280,335] is a movement disorder characterized by unexpected, brief, brisk, shock-like, involuntary, repetitive, synchronous or asynchronous contractions of a muscle or group of axial or appendicular muscles. These involuntary movements may be sufficiently forceful to displace the affected part of the entire body. Myoclonus may occur in combination with dystonia (myoclonic dystonia) [279].
Myoclonus may be focal, multifocal, or generalized. For example, diaphragmatic myoclonus (diaphragmatic flutter) is a rare focal myoclonus causing repetitive, involuntary contraction of the diaphragm and other inspiratory muscles [68]. Myoclonus may occur spontaneously or on attempted movement (action myoclonus) [206] and may be precipitated by cutaneous, auditory, visual, or muscular (e.g., sudden muscle stretch) stimuli. Action or intention myoclonus is most often encountered after cerebral hypoxia (Lance-Adams syndrome) and with certain degenerative disorders, such as Ramsay Hunt syndrome. Myoclonus is seen with structural or metabolic lesions of the spinal cord, brainstem, cerebellum, and cerebral cortex or in normal individuals (e.g., “sleep starts”). Rhythmic myoclonus is typically due to structural lesions of the brainstem or spinal cord. Myoclonus has a relationship to seizures in that both appear to be the result of hyperexcitable neurons. Marsden et al. [243] divide myoclonus into four major etiologies: (a) physiologic, (b) essential, (c) epileptic, and (d) symptomatic. Caviness classified myoclonus as outlined in Table 19.4 [61].
Physiologic myoclonus occurs in neurologically normal individuals. Sleep is the most common circumstance of physiologic myoclonus. The two physiologic forms of myoclonus during sleep or sleep transitions include partial myoclonic jerks (“physiologic fragmentary myoclonus”), consisting of small, multifocal jerks maximal in the hands and face but present diffusely, and massive myoclonic jerks (hypnic jerks). Partial myoclonic jerks are usually multifocal and occur in distal limb muscles, whereas hypnic jerks are generalized and affect the trunk and proximal limbs. Pathologic types of myoclonus that may occur during sleep include isolated periodic movements in sleep, restless legs syndrome with periodic movements in sleep, and excessive fragmentary myoclonus in non–rapid eye movement (REM) sleep. Myoclonus with epilepsy, intention, myoclonus associated with semi-volitional movements, and segmental myoclonus also occur in sleep, but are not primarily nocturnal. Periodic movements of sleep (PMS) or periodic limb movement disorder (PLMD) occurred virtually in all groups of patients referred to a sleep disorder laboratory and consist of repetitive, stereotyped dorsiflexion of the toes and foot, and, occasionally, flexion of the knee and hip. Nocturnal myoclonus often occurs in association with restless legs syndrome. PMS can be asymptomatic for the patient, although, as with all types of nocturnal myoclonus, the disorder may cause distress to the patient’s spouse. On some occasions, however, PMS can induce sleep fragmentation and excessive daytime sleepiness.
Essential myoclonus occurs without any apparent etiology or associated gross neurologic deficit and is characterized by onset before the age of 20 years, sporadic occurrence or dominant inheritance with variable severity, a benign course compatible with an active life and normal life span, absence of other neurologic deficits, and normal EEG [61]. Essential myoclonus is usually distributed throughout the upper body, is exacerbated by muscle activation, and is often responsive to small amounts of alcohol [202]. Some of these patients may exhibit elements of dystonia. Mutations in the gene encoding epsilon-sarcoglycan may cause the myoclonus-dystonia syndrome [372].
Alvarez and Caviness recently described a syndrome of primary progressive myoclonus of aging (PPMA) with the following criteria: (a) asymmetric symptomatic action myoclonus, (b) 65 years of age or older (c) cortical myoclonus physiology, (d) no dementia, (e) no associated features of defined neurodegenerative disorders, and (f) no secondary cause found [8]. In a review of seven patients with this entity, age at presentation ranged from 70 to 87 years with the mean duration from myoclonus onset to last follow-up was 2.9 years. PPMA is a unique syndrome with characteristic findings that differentiate it from dementias and defined neurodegenerative syndromes [8].
Epileptic myoclonus refers to the presence of myoclonus in patients with epilepsy. The myoclonus can occur as only one component of the seizure, the only seizure manifestation, or one of multiple seizure types within the epileptic syndrome. The myoclonus is here presumed to be of cortical origin.
TABLE 19.4 Classification of Myoclonus

Related posts:
 Cranial Nerves IX and X (The Glossopharyngeal and Vagus Nerves)
Cranial Nerve VIII (The Vestibulocochlear Nerve)
The Anatomic Localization of Lesions in the Thalamus
The Localization of Lesions Affecting the Hypothalamus and Pituitary Gland
The Localization of Lesions Affecting the Ocular Motor System
Visual Pathways
Cranial Nerves IX and X (The Glossopharyngeal and Vagus Nerves)
Cranial Nerve VIII (The Vestibulocochlear Nerve)
The Anatomic Localization of Lesions in the Thalamus
The Localization of Lesions Affecting the Hypothalamus and Pituitary Gland
The Localization of Lesions Affecting the Ocular Motor System
Visual Pathways



