The Anatomic Localization of Lesions in the Thalamus
Functional Anatomy of the Thalamus
The paired thalamic nuclei are egg-shaped structures of gray matter located on both sides of the third ventricle [92]. They represent the largest portion of the diencephalon; other diencephalic structures are the epithalamus (pineal and habenular complex), the subthalamic nucleus, and the hypothalamus. The thalamus plays a major role in cortical activation [105]. It projects to the cortex in a highly organized fashion. Characteristically, thalamic connections are reciprocal, that is, the target of the axonal projection of any given thalamic nucleus sends back fibers to that nucleus. Nevertheless, thalamocortical projections are often larger than their corticothalamic counterparts (e.g., the geniculocalcarine projection). Anatomically and functionally, four regions can be distinguished in the thalamus: anterior, posterior, medial, and lateral, partially separated from each other by white matter laminae that are visible to the naked eye [74,132]. In the core of the thalamus, a fifth region is encased by these laminae, the intralaminar nuclei [194]. Finally, the lateral aspect of the thalamus is covered by a layer of myelinated axons, the external medullary lamina, housing in its core clusters of cells, the reticular nucleus of the thalamus [182]. A simplified account of the thalamic nuclei and their connections, pertinent to clinical localization, is given in Table 18.1. Figure 18.1 illustrates the position and main cortical projections of the thalamic nuclei.
From a diagnostic standpoint, the complex thalamic anatomy can be divided into four main regions:
1. The midline, intralaminar, reticular, and some areas of the ventral anterior nuclei mediate general cortical alerting responses and are termed nonspecific thalamic nuclei. However, the location and role of thalamic cells providing nonspecific cortical activation is still being worked out [37,79]. By contrast, specific thalamic nuclei receive sensory information from the body, process it, and project the pertinent output to specific areas of the cortex, such as the somatosensory area and visual cortex. The nonspecific thalamic nuclei receive strong projections from the midbrain reticular formation, hypothalamus and the spinothalamic tract as well as from other sensory pathways. Some stimuli (e.g., auditory, pain) excite this alerting system more easily than others (e.g., visual). These nuclei project back to the midbrain and to the specific thalamic nuclei. Lesions that involve these structures bilaterally cause impairment of alertness.
2. The medial (dorsomedial) and anterior thalamic nuclear groups play an important role in memory and emotions [97]. They are connected with the hypothalamus, the “limbic lobe” (cingulate gyrus, medial temporal region, insula), and the frontal lobe. The dorsomedial nucleus mediates olfaction, emotions, the secondary affect of pain, the sleep-wake cycle, and executive functions [7,156,163,170,187,190]. Lesions of the anterior nucleus are more consistently associated with memory and executive function loss [55,63,69].
3. The ventral lateral and basal nuclear groups are concerned with the processing of sensory information and relaying it to the cortex, and with sensorimotor control [108].
A. Relaying sensory information to the cortex is effected mainly by three nuclear groups, as follows:
i. The ventral posterior nuclear group, where taste and somatosensory information is elaborated and projected to the somatosensory cortex of the parietal lobe. Within the thalamus, lateral inhibition increases sharpness in spatial localization. Information from receptors in the head reaches the ventral posterior medial nucleus through the trigeminothalamic pathways. The ventral posterolateral nucleus processes somatosensory information conveyed by the spinothalamic tract and medial lemniscus. A group of nuclei, dorsal to the medial geniculate body and called the ventral medial or caudal nucleus, participate in the perception of pain and temperature, particularly well-localized, sudden pain [12]. Discrete spinothalamic projections also reach other nuclei, including the ventrolateral nucleus [35].
TABLE 18.1 Source and Destination of Thalamic Connectionsa
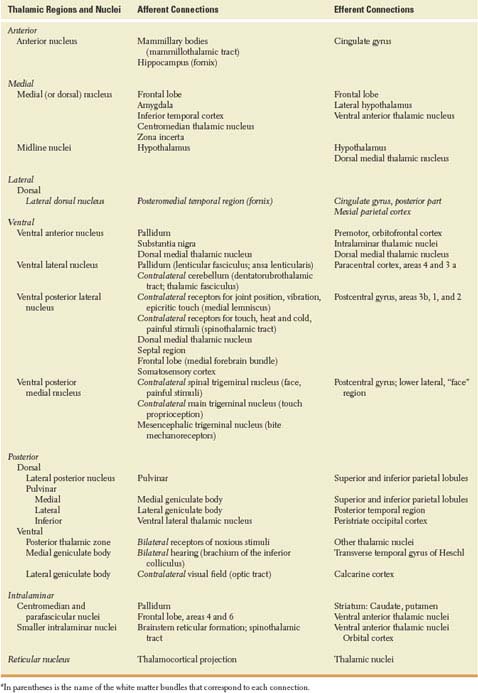
ii. The medial geniculate body, where auditory information from the inferior colliculus passes on to the transverse temporal gyrus, buried in the depth of the Sylvian fissure. Lesions of this thalamic nucleus are discussed in Chapter 11.
iii. The lateral geniculate body, relay station for the visual pathway, which receives from the retinal ganglion cells the axons that form the optic tract and which originates the axons that project to the calcarine cortex through the optic radiations. Lesions of this thalamic nucleus are discussed in Chapter 7.
B. Sensorimotor control is carried out by the more anterior of the ventrolateral nuclei (ventral lateral and ventral anterior) and perhaps by the intralaminar nuclei [93,124].
iv. The ventrolateral nucleus has an oral, a medial and a smaller caudal portion. The medial portion receives input from the cerebellum and projects to the precentral cortex or primary motor cortex (area 4). The largest oral portion receives input from the globus pallidus and projects mostly to the premotor cortex. The ventrolateral nucleus also receives information from musculoskeletal system mechanoreceptors; it contributes to the coordination of finer, distal motor movements with the proximal axial movements that support them.
v. The ventral anterior nucleus may play a role in voluntary attention. It has strong connections with the pallidum, medial thalamic nuclei, and frontal cortex.
Anatomically, the thalamus constitutes the keystone for two large sensorimotor control loops: (a) the cerebello-rubro-thalamo-cortico-pontocerebellar loop and (b) the cortico-striato-pallido-thalamo-cortical loop. Although the physiologic role of each loop is far from clear, it is known that lesions in each loop cause different syndromes. Obviously, the anatomy allows ample possibilities for lesions to affect both loops. Symptoms and signs derived from cerebellar lesions are discussed in Chapter 16. Those derived from basal ganglia lesions are discussed in Chapter 19. Motor findings that point to the thalamus as the site of the lesion are given preferential attention here.
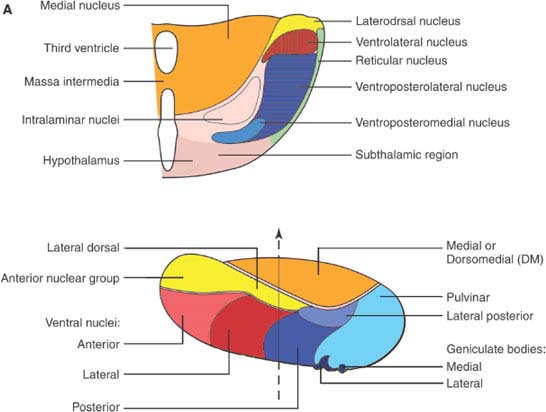
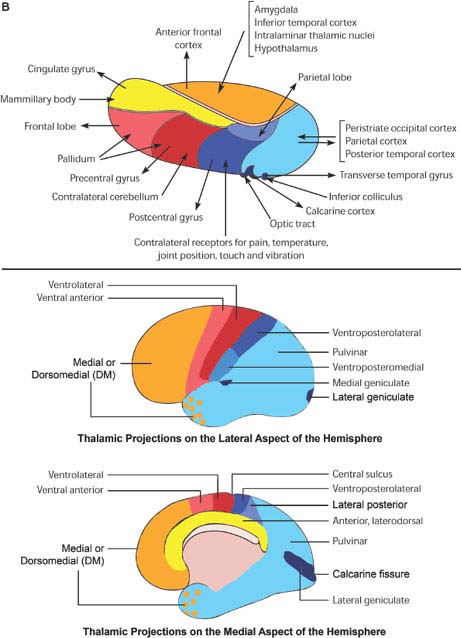
FIG. 18.1. A: Thalamic nuclei. Top, Frontal section of the thalamus at the level of the dashed line on the figure at the bottom. Bottom, Laterodorsal view of the thalamus, showing the position of the thalamic nuclei. (continued)
4. The fourth main region of the thalamus comprises the dorsolateral and posterior nuclear groups, particularly the pulvinar, which seem to modulate occipito-temporo-parietal cortical attention, using an object-based frame of reference [204]. As such, it facilitates visual attention and the cortical attention needed for language- related sensory tasks in the left hemisphere and visuospatial tasks in the right [36,78]. This area is much better developed in humans than in lower mammals. During ontogenesis, it is the last thalamic region to reach adult morphology.
Vascular Supply of the Thalamus
Cerebrovascular disease is the most common cause of discrete thalamic pathology resulting in signs and symptoms of localizing value [165]. Infarcts are more common than hemorrhages. Therefore, some knowledge of the vascular supply of the thalamic nuclei helps greatly to understand the so-called thalamic syndromes and the localization of thalamic lesions.
The thalamic arteries arise from the posterior communicating arteries and from the perimesencephalic segment of the posterior cerebral arteries [25,147–149]. The origin and territory of supply of the various thalamic vessels differ in each person. For instance, when the posterior communicating artery is small or absent, arterial twigs from the posterior cerebral artery supply the thalamic territory that is otherwise supplied by branches of the posterior communicating artery. Table 18.2 summarizes the more common vascular patterns (Fig. 18.2). For this account, the segment of the posterior cerebral artery proximal to the ostium of the posterior communicating artery has been termed the basilar communicating artery [25].
Localization of Ischemic Thalamic Lesions
In localizing ischemic lesions of the thalamus, several points should be kept in mind:
TABLE 18.2 Vascular Supply of the Thalamus
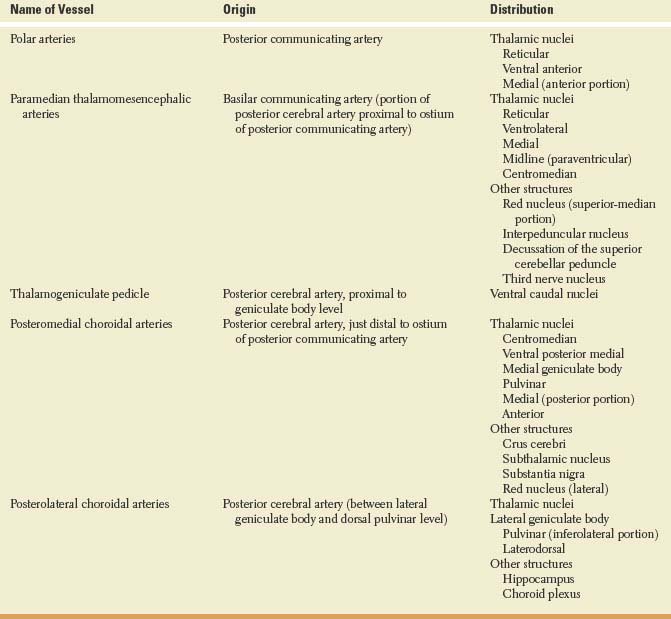
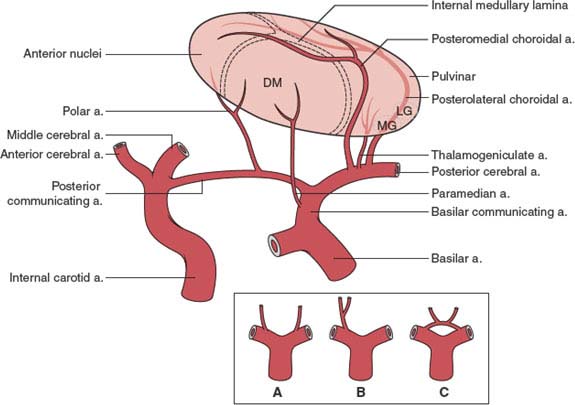
FIG. 18.2. Arterial supply of the thalamus. Inset: Variations in the origin of the paramedian arteries, which may arise from each basilar communicating artery (A), from a single pedicle originating in one basilar communicating artery (B), or from a vascular arcade connecting both basilar communicating arteries (C). DM = dorsomedial; LG = lateral geniculate; MG = medial geniculate. (Modified from Castaigne P, et al. Paramedian thalamic and midbrain infarcts: Clinical and neuropathological study. Ann Neurol 1981;10:127.)
1. The arterial supply for most of the thalamus arises from the vertebrobasilar system, in some cases with a small contribution from the posterior communicating artery [25]. Only the lateral portion and the hilum of the lateral geniculate body are usually fed by the anterior choroidal artery, a branch of the internal carotid artery.
2. Except for the lateral geniculate body, the middle cerebral and anterior choroidal arteries do not supply the thalamus to such an extent that thalamic infarction would result from occlusion of these vessels [148]. Some internal capsular dysfunction, however, may result from occlusion of thalamic vessels [130]. In rare cases the anterior cerebral artery may contribute to the supply of the posterolateral thalamus through a posterior pericallosal vessel, which is anastomotic with the posterolateral choroidal artery.
3. The paramedian thalamic vessels often arise from a single pedicle that originates in one of the basilar communicating arteries (see Fig. 17.2). Thus, unilateral posterior cerebral artery occlusions may result in bilateral paramedian thalamic infarcts [25].
4. The individual pattern in vessel distribution and size will ultimately dictate the presence of one of the syndromes described below.
The arterial territory responsible for a thalamic ischemic infarct may be inferred from the clinical findings, as follows.
Paramedian Territory
Infarcts here tend to involve also the paramedian region of the midbrain. Most often, the syndrome is composed of the clinical triad of somnolent apathy, memory loss, and abnormalities of vertical gaze [73]. Bilateral medial thalamic infarcts account for the behavioral syndrome, and lesions in the area of the rostral interstitial nucleus of the medial longitudinal fasciculus account for the vertical gaze palsy [207]. The following findings may result, depending on the extent and location of the lesion [11,15,25,61,62,65,87,183,207]:
1. Transient loss of consciousness or somnolence; occasionally akinetic mutism
2. Behavioral changes (confusion, agitation, aggression, lack of initiative, disorientation, apathy, manic delirium, a frontal lobe-like syndrome [13])
3. Recent memory loss (with anterograde and retrograde components). Persistent memory loss is observed only with damage of the dominant anterior nucleus or mamillothalamic tract [183]
4. Vertical gaze and convergence disorders (and occasionally blepharospasm)
5. Contralateral hemiataxia, asterixis, or motor weakness
6. Delayed action tremor (occasionally myoclonus or athetosis) in the contralateral limbs
This syndrome is often due to embolic occlusion of the top of the basilar artery or local atheroma at the origin of the posterior cerebral artery [15,20,121,183].
Thalamogeniculate (Lateral Thalamic or Inferolateral Thalamic) Territory
Ischemia in this territory (ventral posterior nucleus, ventral lateral nucleus, and subthalamic region) causes some of the components of the classic thalamic syndrome described by Dejerine and Roussy [15,21,61,62,115,116,122,177,209]:
1. Hemianesthesia (occasionally, proprioception is spared)
2. Transient slight hemiparesis
3. Hemiataxia
4. Hemiataxia–hypesthesia syndrome
5. Lack of nonvolitional utilization of the contralateral body (damaged “automatic pilot”)
6. Dysequilibrium (“thalamic astasia”)
7. Choreoathetoid movements
8. Athetoid posture (“thalamic hand”)
9. Paroxysmal pain (thalamic pain)
10. Homonymous hemianopia (often due to simultaneous medial occipital infarction)
All these findings occur on the side of the body contralateral to the lesioned thalamus. The more severe forms of the syndrome (complete geniculothalamic infarct) accompany proximal occlusion of the posterior cerebral artery [61]. Partial forms (partial geniculothalamic infarct) result from lacunar infarction restricted to one of the penetrating thalamogeniculate vessels [21,61,129] and result in pure sensory or sensorimotor stroke. Disease of such small perforating arteries often accompanies diabetes and chronic hypertension.
Isolated hemiataxia and ipsilateral sensory loss (the hemiataxia–hypesthesia syndrome or thalamic ataxia syndrome) may occur with infarction in the thalamogeniculate territory that involves the lateral part of the thalamus (ventral posterior nucleus and ventral lateral nucleus) [122,177]. The sensory disturbance may be purely subjective, may affect light touch, pain, and temperature sense, or affect light touch, pain, temperature, position, and vibration sense. The contralateral “cerebellar” dysfunction and sensory loss is due to a lesion of the dentatorubrothalamic and ascending sensory pathways into the thalamus [177]. Also, recurrent pure sensory transient ischemic attacks (transient hemihypesthesia) may occur with ventroposterolateral nucleus ischemia [47].
Tuberothalamic (Anterolateral Thalamic) Territory
Infarcts in this territory are due to thalamopolar artery lesions and result primarily in neuropsychological dysfunction [55,61–63]. The following findings may result:
1. Apathy and verbal perseveration, as part of executive function impairment
2. Anterograde memory loss
3. Facial paresis for emotional movement
4. Occasionally, hemiparesis and visual field defects (sensation spared)
5. The superimposition of temporally unrelated information
6. Dysphasia with left-sided lesions
7. Hemineglect and impaired visuospatial processing with right-sided lesions
Bilateral polar artery thalamic infarcts result in apathy, abulia, “frontal lobe” deficits, lethargy, and impaired memory [82].
Territory of the Posterior Choroidal Arteries
These vessels supply the lateral geniculate body, pulvinar, posterior thalamus, and a small posterior portion of the hippocampus and parahippocampal gyri [137]. In lateral posterior choroidal artery territory infarction, the most common clinical manifestations include:
1. Homonymous quadrantanopsia, superior or inferior, or, rarely but particularly suggestive of involvement of the lateral geniculate body in this territory, a homonymous horizontal sectoranopia, tubular or shaped like a wedge
2. Decreased optokinetic nystagmus when moving the drum to the side of the lesion
3. Hemisensory loss with mild hemiparesis
4. Mild hemiparesis, accompanied by sensory loss (although no involvement of the internal capsule is detected by MRI)
5. Transcortical aphasia
Isolated medial posterior choroidal artery territory infarction has not been reliably documented. Miosis, occasionally ipsilateral, has been described with these lesions [137].
Thalamic hemorrhage is discussed in Chapter 21.
Clinical Manifestations of Lesions in the Thalamus
The following considerations facilitate the understanding of the clinical manifestations of thalamic lesions:
1. Because of the smallness of the thalamus, several of the nuclei and even several of the functional regions outlined above are usually affected simultaneously, even by discrete lesions such as infarcts. Because arteriolar vascular territories cross the nuclear boundaries, as a rule ischemic disease affects several nuclei, often partially [25]. In addition, many lesions are not restricted to the thalamus, but involve neighboring areas of the brain as well. Paramedian thalamic vascular lesions tend to affect also the midbrain, with a resultant decrease in the level of alertness to the point of coma [25]. Thus, other motor or sensory findings that would point to thalamic involvement cannot be elicited. Laterally located lesions may disrupt the internal capsule, thereby causing motor and sensory deficits [64] that mask the deficits characteristically present with thalamic involvement. For instance, the tendency to avoid using an otherwise strong limb contralateral to a lesioned ventral lateral thalamic area (“thalamic neglect”) is not manifest if capsular involvement has resulted in a hemiparesis. Lesions extending inferiorly may yield hemiballismus, for which the subthalamic lesion is probably primarily responsible. Lesions in the territory of the lateral posterior choroidal artery may cause memory loss through involvement of the parahippocampal gyrus [137].
2. Except for sensory deficits, unilateral thalamic lesions result in transient deficits. By contrast, bilateral lesions or unilateral lesions, such as hemorrhages or tumors, which press against the contralateral thalamus or impinge on the midbrain, may render the patient comatose or akinetic and mute.
3. Timing has a particular impact on the clinical expression of thalamic lesions. As the effects of an acute lesion recede, neglect may disappear, inability to walk may yield to mild ataxia, and hemisensory loss diminishes. Other findings, however, particularly the so-called positive symptoms (tremor, pain), usually become more pronounced within a few weeks after the injury.
Discrete lesions in various regions of the thalamus, and, more recently, deep brain stimulation through implanted electrodes, are increasingly used for the treatment of parkinsonian and essential tremor [144,199], dystonia [91], pain [34], epilepsy [188], and the manifestations of Gilles de la Tourette syndrome [154,186]. Tremor treatment is the most extensively used and best understood deep brain stimulation thalamic procedure. Essential tremor can be treated by deep brain stimulation with electrodes in the ventrolateral nucleus. The ventrolateral nucleus includes the nuclei ventralis intermedius and ventralis oralis posterior. The ideal location of the stimulating electrodes seems to lie in the ventralis oralis posterior nucleus immediately anterior to the cerebellar receiving area, ventralis intermedius [144]. In addition to the target effect, some of these procedures have produced other symptoms or signs, which will be mentioned under the appropriate heading.
Disturbances of Alertness
Sudden bilateral paramedian thalamic lesions, such as infarcts, may cause a decreased level of alertness ranging from somnolence to coma [25,61,106], generally transient. Prolonged coma may result if the lesion extends into the midbrain tegmentum. These patients often have oculomotor paresis [25]. By contrast, patients with pure thalamic involvement have very small reactive pupils (“diencephalic pupil”), and their extraocular movements, elicited by the doll’s head maneuver, are full [152]. Akinetic mutism, discussed in Chapter 23, may follow bilateral paramedian thalamic lesions [25,171], as in the noted Karen Quinlan case [90].
The intralaminar, reticular, and ventral anterior nuclei seem to play the greatest role in mediating normal alertness [67,182]. Electrical stimulation of this region induces arousal from sleep [208], while lesions here cause drowsiness [71]. Bilateral paramedian thalamic infarction may cause severe apathy and a bromocriptine-responsive compulsive tendency to assume a sleeping posture (“presleep behavior”) [26]. In patients with paramedian thalamic lesions and daytime hypersomia, REM sleep is normal, but wakefulness, sleep spindling and stages of deep sleep are all reduced, suggesting that the medial thalamus is the “final common pathway” for both maintenance of wakefulness and promotion of non-REM sleep [7,174]. Other reported disturbances include inversion of the nycthemeral rhythm and dissociation of sleep stages, detected by electroencephalography, between the two hemispheres: the hemisphere affected by a thalamic tumor showed earlier onset of deeper sleep stages [81]. Normally, thalamic deactivation at sleep onset precedes that of the cerebral cortex [109]. The intralaminar nuclei of the thalamus are involved in the genesis of unconsciousness with seizures [192]. Low-frequency (3/s), high-intensity combined stimulation of the right centromedian nucleus and left nonspecific mesencephalic ascending pathways elicits a response similar to the typical absence attack [197]. The dorsomedial thalamic nucleus may have reduced volume and be hypometabolic on the side of chronic temporal lobe epilepsy [80].
Fatal familial insomnia, a prion disease related to mutations in the PRNP gene on chromosome 20, is mostly confined to the anterior and dorsomedial thalamic nuclei but also involves the intralaminar nuclei [131]. It is characterized by progressive insomnia, loss of slow-wave sleep and abnormal REM sleep behavior, a loss of vegetative and endocrine circadian rhythms, and dysautonomia (hyperhidrosis, hyperthermia, tachycardia, hypertension, miosis, and sphincter disturbances). These disturbances are associated with impaired arousal during daytime, dreamlike states, motor abnormalities (dysarthria, ataxia, pyramidal dysfunction, intention tremor, myoclonus), and eventual coma and death [131].
Autonomic Disturbances
Deep brain stimulation with electrodes in the centromedian-DM thalamic region caused a change in penile erection, facilitating it in a patient and inhibiting it in another [186]. In both these cases deep brain stimulation improved the tics of Gilles de la Tourette syndrome, where an abnormality of anterior thalamic dopamine activity has been detected [180]. Kleine-Levin syndrome, which is characterized by episodes of somnolence, hyperphagia, impaired recent memory, and hypersexual behavior, traditionally believed to be related to hypothalamic disease, may be due to paramedian thalamic lesions [24].
Disturbances of Mood and Affect
Apathy, disinterest, and a lack of drive have been reported with lesions of the paramedian region of the thalamus [13,25,61,87], which is involved in reward learning [160]. Less often, such lesions may cause agitation, dysphoria, or an acute confusional state [13,57], and even undue joviality, accompanied by confabulation [25]. Similar manifestations of bilateral medial thalamic damage may be interpreted as a partial Klüver-Bucy syndrome, described in a patient with chronic amnesia, distractibility, hyperorality, affective dyscontrol, and a socially inappropriate behavior [134]. A manic-like state with disinhibition affecting speech (with logorrhea, delirium, joking, laughing, inappropriate comments, and confabulation) has been described with right thalamic lesions [13,94]. A patient mentioned having lost the pleasure involved in reading after a left anterior thalamic infarct [32]. The ipsilateral cingulate gyrus was hypometabolic. In schizophrenia, a disorder with altered affect and executive function, neuronal loss has been found in the dorsomedial and anterior nuclei of the thalamus by some [153,210], but not by others [44].
Memory Disturbances
Recent memory may be transiently or permanently impaired by lesions of the anterior or medial thalamic nuclear region [25,62,63,76,127,198]. This deficit appears most consistently with bilateral lesions but may be associated with even unilateral lesions of either thalamus [25,32,145]. In some cases, the transient nature of the deficit has prompted the diagnosis of transient global amnesia, although most patients with this disorder do not present evidence of thalamic disease [59,157].
The proposed anatomic basis for a permanent amnestic syndrome after bilateral anterior thalamic infarctions is combined damage to hippocampal– thalamic pathways via the mammillothalamic tract and medial temporal–thalamic pathways via the inferior thalamic pedicle [63,68,110,195,200]. These pathways are closely adjacent in the anterior thalamus, and bilateral lesions that cause amnesia are found in this region, whereas bilateral medial thalamic lesions that do not cause amnesia are located more posteriorly [63,110,125,200]. Korsakoff’s amnesia correlates with neuronal loss in the anterior thalamic, but not dorsomedial, nuclei [69]. Pure amnesia has also been described after a unilateral left polar thalamic infarct affecting the anterior thalamic nuclei and adjacent mammillothalamic tract [32]. Lesions involving the left thalamus affect mainly verbal memory [55,133], whereas those in the nondominant paramedian thalamic region impair memory related to visuospatial tasks (nonverbal memory) [13,150,178,179,206].
Thalamic amnesia is characterized by deficits in anterograde verbal and visual learning and in retrograde amnesia, but motor learning is preserved [63]. Patients who are alert and active usually perform adequately in tests of immediate memory, such as digit span. Characteristically, the amnesia is most profound for events taking place after the injury (anterograde amnesia or recent memory loss), although sometimes it includes information acquired from days to years previously (retrograde amnesia) [55,83,120,211]. Disorientation to time is common. With thalamic lesions, the content of recall is not as affected as the temporal order of the items stored in memory, be they verbal or nonverbal items. Thus, the patients may retrieve facts, but in a disorganized fashion and out of context [55,175]. Some patients seem to be aware of their deficit [211], and others do not [198]. Whether this can be accounted for by differences in the site or extent of the lesion remains to be determined.
Some unusual patterns of memory loss and recovery have been described with thalamic lesions. A patient with bilateral medial thalamic lesions recognized by their voices relatives whom he had failed to identify visually (prosopagnosia) [120]. Another patient’s retrograde amnesia improved suddenly one year after a left anterior thalamic infarction, when he was exposed to an event that triggered the recall of a flood of previously forgotten autobiographical detail [107]. The resemblance with Proust’s recollection triggered by the taste of aunt Leonie’s “petite madeleines” has led to the term “petite madeleines phenomenon” for this remarkable presentation [107].
Confabulation, or falsification of memory occurring in clear consciousness, is frequently present with thalamic amnesia [13,55,167,168]. Patients may confabulate spontaneously or when asked to recall some facts [167,168]. Particularly those that confabulate spontaneously seem to have an impaired ability to order in time facts retained in memory [55,168]. Part of the problem has to do with identifying the relevance of items stored in memory to the current context. Items of memory that are not appropriate for the here and now find their way into the patient’s verbal output or are manifested by behavior that relates to past experiences, but not to what is now appropriate.
Sensory Disturbances
Thalamic lesions may cause sensory loss, often accompanied by paresthesias and pain.
PARESTHESIAS AND PAIN
Clinically, small lesions in the ventral posterior lateral nucleus of the thalamus may yield only contralateral paresthesias that lack “objective” sensory loss when tested at the bedside [49]. Such paresthesias tend to occur on one side of the face, particularly around the mouth, and in the distal portion of the limbs. Occasionally, this cheiro-oral or distal distribution of the paresthesias may suggest a more distal lesion (e.g., radiculopathy) [98]. These areas of the body have the largest representation in the thalamic sensory nuclei. When the trunk is also numb, the subjective feeling of numbness may stop abruptly in the midline, although on objective testing the sensory loss often fades toward the midline [130]. Such a “thalamic midline split,” which is absent with parietal lesions, has been thought to have some clinical value in identifying the site of the lesion [129]. The numb areas of the body may feel swollen, enlarged, shortened, twisted, or torn, or they may tingle. Objects held with the limb contralateral to the lesion may feel abnormally heavy. Finally, the patient may be unaware of a profound sensory loss.
Pain referred to as thalamic pain is perhaps the best known component of Dejerine and Roussy’s thalamic syndrome, described above [119,209]. The unpleasant or excruciatingly painful sensation on the side of the body contralateral to a thalamic lesion (an infarct is most common) may appear at the time of the injury [49] or when the sensory loss begins to improve. The pain feels localized to the skin. Cutaneous stimuli trigger paroxysmal exacerbations of the pain, which persists after the stimulus has been removed. The latency between the stimulus and pain perception is prolonged, suggesting that the pathways conveying it are polysynaptic. Because the perception of epicritic pain, such as that induced with a pin-prick, is reduced on the painful areas, this symptom has been termed anesthesia dolorosa, or painful anesthesia. A similar symptom may follow damage of the posterior root ganglia (herpes zoster) or trigeminal nerve or nucleus (trigeminal neuralgia with anesthesia). Ventral-posterior thalamic nuclear lesions are more likely to produce half-body pain than lesions elsewhere in the sensory pathways [17]. The lesion may be restricted to the ventral-posterior nucleus (ventral caudal nucleus in Schaltenbrand’s terminology [164]) and it is generally accompanied by hypesthesia to cold but not to heat [88]. Metabolic studies have shown enhanced activation of somatosensory cortex with stimulation [88] or pain relief correlating with increased metabolic rates in prefrontal and anterior insular cortices, hypothalamus and periaqueductal gray [95], all of them structures felt to play an important role in pain perception. Some patients with localized neuropathic pain can be relieved by stimulation of the basal ventroposteromedial region of the thalamus or by lesioning the sensorimotor cortex [88].
Thalamic pain seldom occurs with tumors. It has been described most often with vascular lesions, some of which involve not only the thalamus but also the deep parietal white matter [2]. Besides, delayed pain may follow cortical parietal infarcts, particularly those in the bank of the Sylvian fissure, affecting the second somatosensory area (pseudothalamic syndrome) [10,166].
LOSS OF SENSORY MODALITIES
All somatosensory modalities are processed in the ventral posterior nucleus of the thalamus contralateral to the side of the body where they are perceived. Within the nucleus there is a definite topographic distribution: the head is represented anteroinferomedially, whereas the leg is represented posterosuperolaterally; the arm is represented in an intermediate position. A larger volume of the nucleus is dedicated to the mouth, tongue, and distal portion of the extremities; their thalamic representation is almost completely crossed. The large oral thalamic and cortical representation in humans may well be related to language functions [27]. The face, proximal portion of the limbs, and trunk are represented in a smaller volume of thalamic tissue, mainly contralateral but partially ipsilateral [18]. Thalamic sensory loss tends to occur maximally in the distal portion of the limbs and often spares the face [89]. Such sparing may be related to the different vascular supply of this portion of the ventroposterior region (paramedian territory) or to the bilateral thalamic representation of the face.
In regard to the thalamic topography of various sensory modalities, physiologic experimental studies have shown that cells concerned with deep pressure and movements of the limbs are preferentially located in the rostral and caudal ends of the ventral posterior lateral nucleus. The central part of the nucleus contains neurons that respond to cutaneous stimuli. In humans, however, lesions large enough to produce any sensory loss most often involve several modalities. No existent clinicopathologic studies allow precise identification of thalamic areas for touch versus joint position or vibration. Pain sensation has been obtained by stimulation of the basal part of the nucleus [18].
Because the perception of pin-prick, temperature, touch, or vibration is altered more often after thalamic than after cortical lesions, these sensory modalities have been termed primary or thalamic. By contrast, conscious joint position identification, two-point discrimination, stereognosis, and graphesthesia tend to be more impaired after cortical parietal lesions, and are thus termed secondary or cortical sensory modalities. Nevertheless, parietal lesions often cause some impairment of thalamic modalities and vice versa. Occasionally, a lesion in the thalamus may disturb mainly the so-called cortical sensory modalities [51,203].
Anesthesia and impaired temperature perception tend to occur with basal lesions near the medial geniculate body [17,18]. Because vibration sense remains unaltered after surgical removal of the parietal cortex, it has been assumed that hemispheric lesions causing loss of vibratory sense necessarily implicate the thalamus or the thalamocortical projections [161].
Decreased thalamic perfusion has been observed with hysterical hemisensory loss [201]. This abnormality reverted when the patients improved.
Disturbances of vision and hearing are discussed in Chapters 7 and 11. Visual field defects caused by thalamic lesions frequently involve the superior quadrant bilaterally. An intolerance to light (“central dazzle”) has been ascribed to a thalamic lesion [38,42]. Paramedian thalamic infarction may cause the sudden onset of vivid, formed visual hallucinations (suggesting peduncular hallucinosis) associated with agitation and sleep disturbance (MRI revealed no abnormality of the midbrain or cerebral peduncles) [46]. Vivid visual hallucinations, suggesting peduncular hallucinosis, with left hemiparesis and left paresthesias have been described with a right posterior thalamic infarct [173]. Auditory and visual experiential hallucinations may occur with unilateral thalamic lesions affecting the intralaminar and dorsomedial nuclei [112,139]. Auditory illusions of hyperacusis and palinacousis may occur with a lesion in the medial geniculate body [52]. Auditory-tactile synesthesia followed a lesion centered in the ventrolateral nucleus [9]. Unilateral visual sensory neglect may occur with lesions of the right pulvinar [85,193].
Normal detection, but altered identification, of smells has been associated with thalamic lesions; in several patients, odors and taste were perceived either in a neutral way, their pleasant character having disappeared, or as unpleasant [162,172]. Taste sensations have been elicited in humans by stimulation of a portion of the ventro-postero-medial nucleus [101].
Motor Disturbances
Just as the sensory disturbances described in the previous section can be related to lesions in the ventral posterior nucleus of the thalamus, motor disturbances can be related to lesions of the ventral lateral nucleus and the adjacent subthalamic region.
POSTURAL DISTURBANCES
Following an acute thalamic lesion, even a unilateral lesion, patients may be transiently unable to stand or even sit, despite normal strength of the limbs when tested against resistance (thalamic astasia) [115,116,196]. The lesions, including infarction, hemorrhage, or tumor, primarily involved the superoposterolateral thalamus and spared the rubral region. Although alert, with normal or near-normal strength and a variable degree of sensory loss, patients with thalamic astasia cannot stand, and often cannot even sit up unassisted. They fall backward or toward the side contralateral to the lesion and appear to have a deficit of overlearned motor activity of an axial and postural nature [116]. Sudden falling to one side while sitting, standing, or walking has also been described with basal ganglia lesions contralateral to the side of the fall [96]. Some patients with posterolateral thalamic lesions push actively to the side contralateral to the lesion (pusher syndrome) [86]. The postural disturbance of thalamic lesions may be accompanied by a disturbance in the patient’s perception of the vertical axis [39,40,84].
Postural abnormalities in patients with lesions in the ventrolateral nucleus or its connections with the medial frontal region, in the suprathalamic white matter, go beyond simple disequilibrium [115,116]. Volitional movements, like trying to overcome the strength of the examiner during isometric testing, are normal. Yet the patient does not use the same strong limbs, and particularly the axial muscles, in tasks that are normally performed automatically or without much thinking, such as shifting in bed. Proximal movements that normally support distal ones, such as abduction of the shoulder when trying to pick up a cup, are restricted, even though the patient is perfectly able to abduct his or her shoulder on command. This syndrome occurs with lesions of either thalamus and is different from the sensory hemineglect described predominantly with nondominant thalamic lesions [85,116]. It has been called motor neglect [111]. Neglect to use the limbs contralateral to the lesion may convey to the examiner the false impression that the patient is hemiplegic [6,13,15,146,205,206]. Certainly large infarcts, lacunes [129], hematomas [203], and tumors may involve the neighboring internal capsule, causing a more or less profound hemiplegia. However, purely thalamic involvement does not result in hemiparesis. Lesions in the ventral lateral nucleus of the thalamus cause contralateral hypotonia, reduction of emotional expression, and transient neglect [71,111]. This syndrome may occur even with lesions that are discrete enough to spare all sensory modalities and the early (thalamic) components of the somatosensory evoked response. Such lesions, which are circumscribed to the ventral lateral nucleus, spare the ventral posterior nucleus. Because the late components of the somatosensory evoked response are abolished, it has been postulated that the ventral lateral nucleus plays a key role in the activation of the frontal cortex. Unilateral thalamic lesions cause akinesia, either as a result of sensory inattention or as a consequence of impaired activation of axial, automatic synergies, as described above [15,115,196,206].
Lesions of the thalamus may cause emotional facial paresis (i.e., weakness of emotionally evoked facial movements, such as smiling, with normal volitional activation) [75]. Contralateral emotional facial paresis has been described with lesions of the thalamus and subthalamus, anterolateral thalamus and insula, posterior thalamus and operculum, and posterior thalamus [14,15,62,75].
Damage to the dentatorubrothalamic projection to the ventral lateral nucleus by a lesion rostral to the decussation of the superior cerebellar peduncle or damage to the ventral lateral nucleus itself results in hemiataxia (coarse, with action tremor, dysmetria, dysdiadochokinesia, and rebound) of the contralateral limbs [113]. These structures are near the corticospinal pathways and the ventral posterior nucleus of the thalamus, explaining why the hemiataxia is associated with hemiparesis or hypesthesia in this type of infarct [123]. Isolated hemiataxia and ipsilateral sensory loss (the hemiataxia-hypesthesia syndrome) may be a manifestation of thalamic infarction in the thalamogeniculate territory causing damage to the ventral posterior nucleus and ventral lateral nucleus [122]. The cerebellar syndrome is not as severe as with involvement of the superior cerebellar peduncle or dentate nucleus. Regarding hand movements, pinching may be involved in both cases, but reaching tends to be spared with thalamic lesions [8]. Some weeks after the injury (which is most often ischemic), tremor at a rate of between three and five cycles per second may appear in the affected extremities [128]. It is mainly distal and increases greatly during the performance of any movement. This tremor may be abolished by DBI or by a surgical lesion of the ventral lateral nucleus of the thalamus [169]. If the central tegmental tract is also involved, tremor with a similar rate may affect the eyelids, eyes, or palate (“palatal myoclonus or tremor”). Central tegmental tract lesions are in the territory of the thalamopeduncular paramedian vessels and are often related to occlusion of the top of the basilar artery [25]. Contralateral cerebellar ataxia and proprioceptive sensory loss may occur with lesions of the ventroposterior thalamus, likely due to interruption of cerebellar outflow pathways in the thalamus rather than to sensory deafferentation [66].
Lesions that are slightly more rostral, involving the subthalamic region and the pallidothalamic projections to the ventral lateral nucleus, may cause transient contralateral hemiballismus [94]. After some days or weeks, the amplitude of the movement decreases and either disappears or adopts a choreic or athetotic pattern.
Dystonia may be secondary to a necrotizing lesion or to degeneration, as with the familial dystonias. Messenger RNA for torsinA, a protein encoded by the gene abnormal in early-onset torsion dystonia, is abundantly present in the thalamus [4]. Thalamic infarcts in the intermediate and caudal portions of the ventrolateral nucleus may cause myoclonic dystonia in the contralateral limbs [54,93,99,103]. The myoclonic nature of the deficit, often accompanied by action tremor or chorea, differs from the dystonia with tonic spasms more characteristic of striatopallidal lesions [93,99]. The onset of the dystonic movements often lags by months or years after the acute insult [54,56]. Dystonic tremor with chronic MRI evidence of infarction in the anterior nucleus of the thalamus may have resulted from a more posteroventral lesion causing atrophy but no cavitary lesions in the involved nuclei [30]. The sensory fields of thalamic neurons are enlarged in patients with dystonia [100,102]. Patients with secondary dystonia or hemiballismus of basal ganglionic origin tend to improve with lesions or stimulation of the ventrolateral nucleus of the thalamus [23].
Abnormal posturing of the hand, often termed thalamic hand, may appear two or more weeks after the occurrence of a vascular lesion of the same region. The hand assumes a posture with flexion at the wrist and metacarpophalangeal joints, whereas the interphalangeal joints are hyperextended. Flexion of the metacarpophalangeal joints increases from the second digit, which may actually be extended, to the fifth digit, which is markedly flexed. The fingers may be forcibly abducted. The thumb is either abducted or pushed against the palm [114].
Other abnormal movements described with thalamic lesions include action myoclonus [5], ideomotor apraxia [136], and asterixis [19,41,116,181]. Asterixis is more common with ventrolateral nucleus lesions [184]. Hyperekplexia, the sudden loss of postural tone caused by startling stimuli, may be exacerbated with thalamic lesions [45]. Imitation synkineses, also called mirror movements, are common after thalamic lesions [117]. In such cases the distal portion of the limb contralateral to the thalamic lesion tends to imitate the movement performed by the healthy side. When the patient forcibly makes a fist with the sound hand, the fingers of the other hand curl up into the palm, and the patient ends by making a fist with both hands. Loss of position sense or loss of cortical activation by the thalamus may underlie these abnormal movements, which can be decreased, like the choreic movements described above, when the patient concentrates his attention on avoiding them.
A decreased corneal reflex may be present in patients with hemiparesis and hemisensory loss due to a cerebral hemispheric lesion [48]. Loss of parietal excitatory influence on the lower brainstem seems to be responsible for this finding [141]. Pure thalamic lesions, even those that cause a marked hemisensory loss, do not depress the corneal reflex [142]. However, a patient with an acute thalamocapsular lesion had bilaterally depressed late components of the blink reflex when the side contralateral to the lesion was stimulated [29]. It is possible that damage of corticopontine fibers traveling in the internal capsule may be responsible for the deficit.
Small surgical lesions or chronic stimulation of the ventrolateral nucleus of the thalamus may ameliorate drug-resistant tremor [169].
Disturbances of Ocular Motility
Lesions restricted to the thalamus cause only subtle changes in ocular motility [143]. Visual information from the superior colliculus, relayed by the pulvinar to the parietal lobe (“second visual system”), contributes to the detection and localization of visual events in space and to the production of saccadic eye movements that allow the “first” (geniculostriate) visual system to identify such events. Lesions in the pulvinar have been said to cause (a) a decrease in the critical flicker frequency and neglect of visual objects in the periphery of the contralateral visual field, (b) prolonged latency of visually evoked saccadic eye movements, and (c) a paucity of spontaneous eye movements directed toward the contralateral hemifield [212].
Much more striking, and more obvious at the bedside, are eye movement abnormalities that occur when a lesion involves the midbrain and thalamus; this often happens with paramedian thalamopeduncular infarcts [25]. In such cases, impairment of ocular motor function results in abnormal pupils, ptosis, and restriction of vertical eye movements and of adduction. Selective upgaze, downgaze, or combined dysfunction may occur [15], as may blepharospasm [155], bilateral internuclear ophthalmoplegia with ptosis [15], and pseudo-sixth nerve palsy [20] (see Chapter 8). Thalamic lesions may be associated with vertical gaze palsies not because of thalamic injury per se but because of extension of the lesions into the upper midbrain [176]. A “vertical one-and-a-half syndrome” (vertical palsy in one eye, upward palsy in the other eye) may occur [14,15,183]. Bilateral medial thalamic lesions may cause purely vertical saccadic apraxia, affecting volitional vertical saccades but not saccades elicited by external stimuli [127]. Acute thalamic esotropia, with impaired upward gaze, has been described with infarction of the contralateral posterior thalamus in the basilar-communicating artery territory [58]. Tonic activation of the medial rectus in this case could result from damage to direct inhibitory projections from the thalamus or impairment of inputs to midbrain neurons involved with vergence control [58].
Large thalamic hemorrhages may impinge on the midbrain or impair its function by causing raised intracranial pressure [57]. The eyes then become tonically deviated down and slightly adducted, as if peering at the tip of the nose [203]. In some instances, the eyes may be tonically deviated to the side of the hemiparesis, opposite a thalamic hemorrhage (“wrong-way eyes”) [50,189]. This finding has also been reported in extrathalamic supratentorial lesions [151,189].
Deep brain stimulation for the treatment of epilepsy, with electrodes placed at the mesodiencephalic junction, just inferior to the centromedian nucleus of the thalamus, may cause nystagmus with constant velocity slow phases, beating to the right when the left thalamus is stimulated and vice versa [185].
Depression of the reticular activating system (most often metabolic in nature) or involvement of both thalami results in small pupils (1 mm in diameter) that react well to light (diencephalic pupils) [152]. Anisocoria is occasionally present, with the smaller pupil ipsilateral to the thalamic lesion.
Disturbances of Complex Sensorimotor Functions
The thalamus modulates the association cortex involved in the processing of language and other “higher” cortical functions. Although compared with large cortical lesions, unilateral thalamic lesions do not impair these functions as much; the pattern of impairment has some localizing value. Mention has already been made of the contralateral motor neglect caused by lesions in the ventral lateral nucleus of the thalamus and of the contralateral visual inattention that results from lesions in the pulvinar.
Patients with right thalamic lesions may have constructional apraxia and display marked neglect of the left hemifield [205,206]. This hemineglect may be associated with anosognosia and asomatognosia, thus mimicking a parietal lobe lesion [13,205], and is likely due to damage to the intralaminar and ventrolateral or ventral anterior nuclei on the right [31,206]. Right thalamic lesions can also cause impairment in the identification of emotional facial expressions with preserved discrimination of facial identity (prosopoaffective agnosia) [202]. Alexia related to impaired visuospatial perception has been described after right thalamooccipital infarction [72]. Optic ataxia or visuomotor apraxia has been described in a patient with CT evidence of bilateral lesions in the pulvinar, the one on the left in the acute stage when the patient was studied [33].
Dominant-hemisphere thalamic lesions may cause a transient language disturbance (thalamic aphasia) [15,60,77,191] characterized by (a) reduced spontaneous speech with paraphasic errors and perseveration, (b) varying degrees of auditory comprehension impairment, (c) preserved repetition and reading, (d) defective spontaneous writing and writing to dictation but normal copying, (e) word-production anomia but spared word selection and word symbolism, and (f) distractibility. This deficit, which would be classified as a mixed transcortical aphasia, tends to improve in a few weeks [3,22,77]. The language deficit may have more semantic components when the lesion is posterior [158] and more motor components when the lesion affects the anterior nucleus [55]. Electrical stimulation of the ventrolateral thalamus produces an acceleration of speaking. The patient feels urged to speak faster [71]. Stimulation also enhances later recall of objects presented to the patient. The left thalamus is involved in attention mechanisms that gate storage and retrieval of both long-term and short-term verbal memory [78]. Derangement of a specific cortical attention mechanism because of the thalamic lesion results in a lack of drive to speak and perseveration of apparently unrelated verbal material [159]. Some patients have hypophonia and dysarthria [55]. Such language impairment resembles the language impairment that results from left medial frontal lesions [16,118]. Other dorsomedial thalamic lesions cause a bizarre language pattern, with some characteristics of dysfunction in the prefrontal cortex. It contains intrusions (segments of speech out of context) and other evidence of temporal gating impairment, such as giving biographical information while working on a calculation test [28,55]. Apraxic agraphia has been described with an MRI showing a dorsomedial thalamic lesion [140].
The language impairment noted with left thalamic lesions and the visual neglect noted with right thalamic lesions are associated with decreased activity of ipsilateral frontal or temporoparietal association cortex [6,126,140,146]. Deafferentation of an otherwise intact cortex (diaschisis) due to the thalamic lesion may explain these “cortical” syndromes with thalamic lesions. For example, thalamic aphasia may be due to decreased cortical activation secondary to the subcortical lesion [6,126,146]. Some authors ascribe to thalamic lesions or thalamocortical disconnection the cases of striatocapsular aphasia reported in the literature [135].
A stuttering-like repetitive speech disorder may occur with infarction in the paramedian thalami and midbrain [1]. Although the speech disorder seems like stuttering, the compulsive repetitions, constant rate, and monotonous tone seen with these infarctions are not associated with ordinary stuttering. The repetitive speech disorder in patients with infarcts in the supplementary motor area have similar clinical features [1].
Disturbances of Executive Function
Paramedian thalamic infarction may cause a lack of initiative and “frontal lobe” utilization behavior (see Chap. 20), suggesting a thalamofrontal component to environmental interactions that requires inhibition, self-monitoring, and cognitive flexibility [43,55,70]. The patients’ language may appear bizarre because of the presence of intrusions (segments of speech out of context) and the superimposition of mental activities normally processed sequentially, for instance, giving biographical information while working on a calculation test [28,55]. Failure of goal-directed regulation of behavior has been observed with a lesion affecting the right anteroventral thalamic region [138]. A left-sided discrete infarction of the medial thalamus may cause severe impairment of complex executive behaviors, probably due to dysfunction of thalamofrontal linkages that help modulate complex human behavior [163]. Repetitive movements (“clonic perseveration”) may be a manifestation of executive function disorder with thalamic lesions [53].
Infarction of the dorsomedial nucleus, intralaminar nuclei, and medial part of the ventrolateral nuclei is often associated with marked hypoperfusion of the overlying frontal region cortex on PET or SPECT [28,70]. It is thought that the “frontal lobe-like” syndrome with thalamic lesions may be related to impaired activation of the frontal lobe by the damaged dorsomedial and anterior thalamic nuclei [13,195], or their projections, for instance with traumatic brain injury [104]. These nuclei provide an important pathway for information from vegetative centers to reach the frontal lobe.
Topographic Localization of Thalamic Lesions
Following the detailed and referenced account of the localization of thalamic symptoms and signs, a brief synopsis follows organized by the anatomical regions of the thalamus.
Anterior Thalamic Region
Discrete lesions may be silent or cause language disturbances when they affect the dominant hemisphere. They may also cause inattention, which results more often when the right hemisphere is involved. Bilateral lesions may cause akinesia, amnesia, and attentional disturbances. Lesions extending to the subthalamic area may cause athetosis, chorea, or postural abnormalities (thalamic hand).
Medial Thalamic Region
Lesions in this location may pass unnoticed when they are small and unilateral. Large or bilateral lesions cause impairment of recent memory, apathy or agitation, attention derangements, and somnolence or coma. Lesions that extend to the midbrain– diencephalic junction may cause contralateral tremor and vertical gaze palsy, affecting particularly downward gaze.
Ventrolateral Thalamic Region
Sensory loss, paroxysmal pains, and hemiataxia in the contralateral side of the body are the most striking sequelae of lesions in the posterior portion of this region. More anterior lesions cause postural abnormalities, such as disequilibrium and restriction of axial supportive movements or delayed tremor. Hemineglect and language disturbances may appear transiently.
Posterior Region
Basal lesions in this region may cause hemianesthesia, “thalamic pain,” and visual field defects. Dorsal lesions give rise to attentional disorders of the ipsilateral hemisphere, resulting in transient aphasia when the dominant hemisphere is involved. Some patients may have myoclonic dystonia.
References
1. Abe K, Yokoyama R, Yorifuji S. Repetitive speech disorder resulting from infarcts in the paramedian thalami and midbrain. J Neurol Neurosurg Psychiatry 1993;56:1024–1026.
2. Agnew DC. Thalamic pain. Bull Clin Neurosci 1984; 49:93–98.
3. Alexander MP, LoVerme SR Jr. Aphasia after left hemispheric intracerebral hemorrhage. Neurology 1980; 30:1193–1202.
4. Augood SJ, Martin DM, Ozelius LJ, et al. Distribution of the mRNAs encoding torsinA and torsinB in the normal adult human brain. Ann Neurol 1999; 46:761–769.
5. Avanzini G, Broggi G, Caraceni T. Intention and action myoclonus from thalamic angioma. Report of a case. Eur Neurol 1977;15:194–202.
6. Baron JC, D’Antona R, Pantano P, et al. Effects of thalamic stroke on energy metabolism of the cerebral cortex. A positron tomography study in man. Brain 1986;109:1243–1259.
7. Bassetti C, Mathis J, Gugger M, et al. Hypersomnia following paramedian thalamic stroke: a report of 12 patients. Ann Neurol 1996;39:471–480.
8. Bastian AJ, Thach WT. Cerebellar outflow lesions: a comparison of movement deficits resulting from lesions at the levels of the cerebellum and thalamus. Ann Neurol 1995;38:881–892.
9. Beauchamp MS, Ro T. Neural substrates of sound-touch synesthesia after a thalamic lesion. J Neurosci 2008;28:13696–13702.
10. Biemond A. The conduction of pain above the level of the thalamus opticus. Arch Neurol Psychiatr 1956; 75:231.
11. Biller J, Sand JJ, Corbett JJ, et al. Syndrome of the paramedian thalamic arteries: clinical and neuroimaging correlation. J Clin Neuroophthalmol 1985;5: 217–223.
12. Blomqvist A, Zhang ET, Craig AD. Cytoarchitectonic and immunohistochemical characterization of a specific pain and temperature relay, the posterior portion of the ventral medial nucleus, in the human thalamus. Brain 2000;123(Pt 3):601–619.
13. Bogousslavsky J, Ferrazzini M, Regli F, et al. Manic delirium and frontal-like syndrome with paramedian infarction of the right thalamus. J Neurol Neurosurg Psychiatry 1988;51:116–119.
14. Bogousslavsky J, Regli F. Upgaze palsy and monocular paresis of downward gaze from ipsilateral thalamo-mesencephalic infarction: a vertical “one-and-a-half” syndrome. J Neurol 1984;231:43–45.
15. Bogousslavsky J, Regli F, Uske A. Thalamic infarcts: clinical syndromes, etiology, and prognosis [published erratum appears in Neurology 1988 August;38(8): 1335]. Neurology 1988;38:837–848.
16. Botez ML, Barbeau A. Role of subcortical structures, and particularly of the thalamus, in the mechanisms of speech and language. Int J Neurol 1971;8:300.
17. Bowsher D, Leijon G, Thuomas KA. Central poststroke pain: correlation of MRI with clinical pain characteristics and sensory abnormalities. Neurology 1998;51:1352–1358.
18. Brodal A. Neurological anatomy in. relation to clinical medicine, 3rd ed. New York, NY: Oxford University Press, 1981:726–754.
19. Calzetti S, Gemignani F, Salati MR, et al. Unilateral asterixis due to thalamic tumor. Case report. Ital J Neurol Sci 1983;4:87–90.
20. Caplan LR. “Top of the basilar” syndrome. Neurology 1980;30:72–79.
21. Caplan LR, DeWitt LD, Pessin MS, et al. Lateral thalamic infarcts. Arch Neurol 1988;45:959–964.
22. Cappa SF, Vignolo LA. “Transcortical” features of aphasia following left thalamic hemorrhage. Cortex 1979;15:121–130.
23. Cardoso F, Jankovic J, Grossman RG, et al. Outcome after stereotactic thalamotomy for dystonia and hemiballismus. Neurosurgery 1995;36:501–507; discussion 7–8.
24. Carpenter S, Yassa R, Ochs R. A pathologic basis for Kleine-Levin syndrome. Arch Neurol 1982;39:25–28.
25. Castaigne P, Lhermitte F, Buge A, et al. Paramedian thalamic and midbrain infarct: clinical and neuropathological study. Ann Neurol 1981;10:127–148.
26. Catsman-Berrevoets CE, von Harskamp F. Compulsive pre-sleep behavior and apathy due to bilateral thalamic stroke: response to bromocriptine. Neurology 1988;38:647–649.
27. Celesia GG. Somatosensory evoked potentials recorded directly from human thalamus and Sm I cortical area. Arch Neurol 1979;36:399–405.
28. Chatterjee A, Yapundich R, Mennemeier M, et al. Thalamic thought disorder: on being “a bit addled.” Cortex 1997;33:419–440.
29. Chia LG. Late blink reflex changes in lesions of thalamus and internal capsule. Neurology 1997;49: 874–876.
30. Cho C, Samkoff LM. A lesion of the anterior thalamus producing dystonic tremor of the hand. Arch Neurol 2000;57:1353–1355.
31. Clark BJ, Bassett JP, Wang SS, et al. Impaired head direction cell representation in the anterodorsal thalamus after lesions of the retrosplenial cortex. J Neurosci 2010;30:5289–5302.
32. Clarke S, Assal G, Bogousslavsky J, et al. Pure amnesia after unilateral left polar thalamic infarct: topographic and sequential neuropsychological and metabolic (PET) correlations. J Neurol Neurosurg Psychiatry 1994;57:27–34.
33. Classen J, Kunesch E, Binkofski F, et al. Subcortical origin of visuomotor apraxia. Brain 1995;118:1365– 1374.
34. Constantoyannis C, Kumar A, Stoessl AJ, et al. Tremor induced by thalamic deep brain stimulation in patients with complex regional facial pain. Mov Disord 2004;19:933–936.
35. Craig AD. Retrograde analyses of spinothalamic projections in the macaque monkey: input to the ventral lateral nucleus. J Comp Neurol 2008;508:315–328.
36. Crosson B. Subcortical mechanisms in language: lexical-semantic mechanisms and the thalamus. Brain Cogn 1999;40:414–438.
37. Crunelli V, Hughes SW. The slow (<1 Hz) rhythm of non-REM sleep: a dialogue between three cardinal oscillators. Nat Neurosci 2010;13:9–17.
38. Cummings JL, Gittinger JW Jr. Central dazzle. A thalamic syndrome? Arch Neurol 1981;38:372–374.
39. Dieterich M, Brandt T. Thalamic infarctions: differential effects on vestibular function in the roll plane (35 patients). Neurology 1993;43:1732–1740.
40. Dieterich M, Bartenstein P, Spiegel S, et al. Thalamic infarctions cause side-specific suppression of vestibular cortex activations. Brain 2005;128:2052– 2067.
41. Donat JR. Unilateral asterixis due to thalamic hemorrhage. Neurology 1980;30:83–84.
42. Du Pasquier RA, Genoud D, Safran AB, et al. Monocular central dazzle after thalamic infarcts. J Neuroophthalmol 2000;20:97–99.
43. Eslinger PJ, Warner GC, Grattan LM, et al. “Frontal lobe” utilization behavior associated with paramedian thalamic infarction. Neurology 1991;41:450–452.
44. Falke E, Han LY, Arnold SE. Absence of neurodegeneration in the thalamus and caudate of elderly patients with schizophrenia. Psychiatry Res 2000;93: 103–110.
45. Fariello RG, Schwartzman RJ, Beall SS. Hyperekplexia exacerbated by occlusion of posterior thalamic arteries. Arch Neurol 1983;40:244–246.
46. Feinberg WM, Rapcsak SZ. ‘Peduncular hallucinosis’ following paramedian thalamic infarction. Neurology 1989;39:1535–1536.
47. Fisher CM. Pure sensory stroke and allied conditions. Stroke 1982;13:434–447.
48. Fisher CM. Some neuro-ophthalmological observations. J Neural Neurosurg Psychiatry 1967;30:383–394.
49. Fisher CM. Thalamic pure sensory stroke: a pathologic study. Neurology 1978;28:1141–1144.
50. Fisher MA, Shahani BT, Young RR. Assessing segmental excitability after acute rostral lesions: II. The blink reflex. Neurology 1979;29:45–50.
51. Friedman JH. Syndrome of diffuse encephalopathy due to nondominant thalamic infarction. Neurology 1985;35:1524–1526.
52. Fukutake T, Hattori T. Auditory illusions caused by a small lesion in the right medial geniculate body. Neurology 1998;51:1469–1471.
53. Fung VS, Morris JG, Leicester J, et al. Clonic perseveration following thalamofrontal disconnection: a distinctive movement disorder. Mov Disord 1997;12:378–385.
54. Ghika J, Bogousslavsky J, Henderson J, et al. The “jerky dystonic unsteady hand”: a delayed motor syndrome in posterior thalamic infarctions. J Neurol 1994;241:537–542.
55. Ghika-Schmid F, Bogousslavsky J. The acute behavioral syndrome of anterior thalamic infarction: a prospective study of 12 cases. Ann Neurol 2000;48: 220–227.
56. Gille M, Van den Bergh P, Ghariani S, et al. Delayed-onset hemidystonia and chorea following contralateral infarction of the posterolateral thalamus. A case report. Acta Neurol Belg 1996;96:307–311.
57. Gilner LI, Avin B. A reversible ocular manifestation of thalamic hemorrhage. A case report. Arch Neurol 1977;34:715–716.
58. Gomez CR, Gomez SM, Selhorst JB. Acute thalamic esotropia. Neurology 1988;38:1759–1762.
59. Gorelick PB, Amico LL, Ganellen R, et al. Transient global amnesia and thalamic infarction. Neurology 1988;38:496–499.
60. Graff-Radford NR, Damasio AR. Disturbances of speech and language associated with thalamic dysfunction. Semin Neurol 1984;4:162.
61. Graff-Radford NR, Damasio H, Yamada T, et al. Nonhaemorrhagic thalamic infarction. Clinical, neuropsychological and electrophysiological findings in four anatomical groups defined by computerized tomography. Brain 1985;108:485–516.
62. Graff-Radford NR, Eslinger PJ, Damasio AR, et al. Nonhemorrhagic infarction of the thalamus: behavioral, anatomic, and physiologic correlates. Neurology 1984;34:14–23.
63. Graff-Radford NR, Tranel D, Van Hoesen GW, et al. Diencephalic amnesia. Brain 1990;113:1–25.
64. Groothuis DR, Duncan GW, Fisher CM. The human thalamocortical sensory path in the internal capsule: evidence from a small capsular hemorrhage causing a pure sensory stroke. Ann Neurol 1977;2: 328–331.
65. Guberman A, Stuss D. The syndrome of bilateral paramedian thalamic infarction. Neurology 1983;33: 540–546.
66. Gutrecht JA, Zamani AA, Pandya DN. Lacunar thalamic stroke with pure cerebellar and proprioceptive deficits. J Neurol Neurosurg Psychiatry 1992;55:854– 856.
67. Hamandi K, Salek-Haddadi A, Laufs H, et al. EEG-fMRI of idiopathic and secondarily generalized epilepsies. Neuroimage 2006;31:1700–1710.
68. Hankey GJ, Stewart-Wynne EG. Amnesia following thalamic hemorrhage. Another stroke syndrome. Stroke 1988;19:776–778.
69. Harding A, Halliday G, Caine D, et al. Degeneration of anterior thalamic nuclei differentiates alcoholics with amnesia. Brain 2000;123:141–154.
70. Hashimoto R, Yoshida M, Tanaka Y. Utilization behavior after right thalamic infarction. Eur Neurol 1995;35:58–62.
71. Hassler R. Thalamic regulation of muscle tone and the speed of movements. In: Purpura DP, Yahr MD, eds. The Thalamus. New York, NY: Columbia University Press, 1966:419–438.
72. Henderson VW, Alexander MP, Naeser MA. Right thalamic injury, impaired visuospatial perception, and alexia. Neurology 1982;32:235–240.
73. Hermann DM, Siccoli M, Brugger P, et al. Evolution of neurological, neuropsychological and sleep-wake disturbances after paramedian thalamic stroke. Stroke 2008;39:62–68.
74. Herrero MT, Barcia C, Navarro JM. Functional anatomy of thalamus and basal ganglia. Childs Nerv Syst 2002;18:386–404.
75. Hopf HC, Muller-Forell W, Hopf NJ. Localization of emotional and volitional facial paresis. Neurology 1992;42:1918–1923.
76. Horel JA. The neuroanatomy of amnesia. A critique of the hippocampal memory hypothesis. Brain 1978;101:403–445.
77. Jenkyn LR, Alberti AR, Peters JD. Language dysfunction, somasthetic hemi-inattention, and thalamic hemorrhage in the dominant hemisphere. Neurology 1981;31:1202–1203.
78. Johnson MD, Ojemann GA. The role of the human thalamus in language and memory: evidence from electrophysiological studies. Brain Cogn 2000;42: 218–230.
79. Jones EG. Viewpoint: the core and matrix of thalamic organization. Neuroscience 1998;85:331–345.
80. Juhasz C, Nagy F, Watson C, et al. Glucose and [11 C] flumazenil positron emission tomography abnormalities of thalamic nuclei in temporal lobe epilepsy. Neurology 1999;53:2037–2045.
81. Kanno O, Hosaka H, Yamaguchi T. Dissociation of sleep stages between the two hemispheres in a case with unilateral thalamic tumor. Folia Psychiatr Neurol Jpn 1977;31:69–75.
82. Kaplan RFea. Bilateral polar artery thalamic infarcts. Neurology 1991;41(Suppl 1):329.
83. Kapur N, Thompson S, Cook P, et al. Anterograde but not retrograde memory loss following combined mammillary body and medial thalamic lesions. Neuropsychologia 1996;34:1–8.
84. Karnath HO, Ferber S, Dichgans J. The neural representation of postural control in humans. Proc Natl Acad Sci U S A, 2000;97:13931–13936.
85. Karnath HO, Himmelbach M, Rorden C. The subcortical anatomy of human spatial neglect: putamen, caudate nucleus and pulvinar. Brain 2002;125: 350–360.
86. Karnath HO, Johannsen L, Broetz D, et al. Posterior thalamic hemorrhage induces “pusher syndrome”. Neurology 2005;64:1014–1019.
87. Katz DI, Alexander MP, Mandell M. Dementia following strokes in the mesencephalon and diencephalon. Arch Neurol 1987;44:1127.
88. Kim JH, Greenspan JD, Coghill RC, et al. Lesions limited to the human thalamic principal somatosensory nucleus (ventral caudal) are associated with loss of cold sensations and central pain. J Neurosci 2007;27: 4995–5004.
89. Kim JS. Pure sensory stroke. Clinical-radiological correlates of 21 cases. Stroke 1992;23:983–987.
90. Kinney HC, Korein J, Panigrahy A, et al. Neuropathological findings in the brain of Karen Ann Quinlan. The role of the thalamus in the persistent vegetative state. N Engl J Med 1994;330:1469–1475.
91. Krauss JK, Yianni J, Loher TJ, et al. Deep brain stimulation for dystonia. J Clin Neurophysiol 2004;21: 18–30.
92. Krauth A, Blanc R, Poveda A, et al. A mean three-dimensional atlas of the human thalamus: generation from multiple histological data. Neuroimage 2010;49: 2053–2062.
93. Krystkowiak P, Martinat P, Defebvre L, et al. Dystonia after striatopallidal and thalamic stroke: clinicoradiological correlations and pathophysiological mechanisms. J Neurol Neurosurg Psychiatry 1998;65:703–708.
94. Kulievsky J, Berthier ML, Pujol J. Hemiballismus and secondary mania following a right thalamic infarction. Neurology 1993;43:1422.
95. Kupers RC, Gybels JM, Gjedde A. Positron emission tomography study of a chronic pain patient successfully treated with somatosensory thalamic stimulation. Pain 2000;87:295–302.
96. Labadie EL, Awerbuch GI, Hamilton RH, et al. Falling and postural deficits due to acute unilateral basal ganglia lesions. Arch Neurol 1989;46:492–496.
97. Lane RD, Reiman EM, Ahern GL, et al. Neuroanatomical correlates of happiness, sadness, and disgust. Am J Psychiatry 1997;154:926–933.
98. Lapresle J, Hagueneau M. Anatomico-clinical correlation in focal thalamic lesions. J Neurol 1973;205:29.
99. Lehericy S, Vidailhet M, Dormont D, et al. Striatopallidal and thalamic dystonia. A magnetic resonance imaging anatomoclinical study. Arch Neurol 1996;53:241–250.
100. Lenz FA, Byl NN. Reorganization in the cutaneous core of the human thalamic principal somatic sensory nucleus (Ventral caudal) in patients with dystonia. J Neurophysiol 1999;82:3204–3212.
101. Lenz FA, Gracely RH, Zirh TA, et al. Human thalamic nucleus mediating taste and multiple other sensations related to ingestive behavior. J Neurophysiol 1997;77:3406–3409.
102. Lenz FA, Jaeger CJ, Seike MS, et al. Thalamic single neuron activity in patients with dystonia: dystonia-related activity and somatic sensory reorganization. J Neurophysiol 1999;82:2372–2392.
103. Lera G, Scipioni O, Garcia S, et al. A combined pattern of movement disorders resulting from posterolateral thalamic lesions of a vascular nature: a syndrome with clinico-radiologic correlation. Mov Disord 2000; 15:120–126.
104. Little DM, Kraus MF, Joseph J, et al. Thalamic integrity underlies executive dysfunction in traumatic brain injury. Neurology 2010;74:558–564.
105. Llinas R, Urbano FJ, Leznik E, et al. Rhythmic and dysrhythmic thalamocortical dynamics: GABA systems and the edge effect. Trends Neurosci 2005;28: 325–333.
106. Lovblad KO, Bassetti C, Mathis J, et al. MRI of paramedian thalamic stroke with sleep disturbance. Neuroradiology 1997;39:693–698.
107. Lucchelli F, Muggia S, Spinnler H. The ‘Petites Madeleines’ phenomenon in two amnesic patients. sudden recovery of forgotten memories. Brain 1995; 118:167–183.
108. Macchi G, Jones EG. Toward an agreement on terminology of nuclear and subnuclear divisions of the motor thalamus. J Neurosurg 1997;86:670–685.
109. Magnin M, Rey M, Bastuji H, et al. Thalamic deactivation at sleep onset precedes that of the cerebral cortex in humans. Proc Natl Acad Sci U S A 2010;107: 3829–3833.
110. Malamut BL, Graff-Radford N, Chawluk J, et al. Memory in a case of bilateral thalamic infarction. Neurology 1992;42:163–169.
111. Manabe Y, Kashihara K, Ota T, et al. Motor neglect following left thalamic hemorrhage: a case report. J Neurol Sci 1999;171:69–71.
112. Manford M, Andermann F. Complex visual hallucinations. Clinical and neurobiological insights. Brain 1998;121:1819–1840.
113. Maraist TAea. Thalamic ataxia. Neurology 1991;41 (Suppl 1):125.
114. Martin JJ. Thalamic syndromes. In: Vinken PJ, Bruyne GW, eds. Handbook of clinical neurology. New York, NY: American Elsevier, 1969:469–496.
115. Masdeu JC, Gorelick P. Impairment of axial and automatic movements with thalamic lesions. Neurology 1989;39(Suppl 1):112.
116. Masdeu JC, Gorelick PB. Thalamic astasia: inability to stand after unilateral thalamic lesions. Ann Neurol 1988;23:596–603.
117. Masdeu J, Rubino F, O’Hara R. Proprioception loss in the genesis of mirror movements. Neurology 1981;31(Suppl 1):89.
118. Masdeu JC, Schoene WC, Funkenstein H. Aphasia following infarction of the left supplementary motor area: a clinicopathologic study. Neurology 1978;28: 1220–1223.
119. Mauguiere F, Desmedt JE. Thalamic pain syndrome of Dejerine-Roussy. Differentiation of four subtypes assisted by somatosensory evoked potentials data. Arch Neurol 1988;45:1312–1320.
120. McEntee WJ, Biber MP, Perl DP, et al. Diencephalic amnesia: a reappraisal. J Neurol Neurosurg Psychiatry 1976;39:436–441.
121. Mehler MF. The rostral basilar artery syndrome: diagnosis, etiology, prognosis. Neurology 1989;39:9–16.
122. Melo TP, Bogousslavsky J. Hemiataxia-hypesthesia: a thalamic stroke syndrome. J Neurol Neurosurg Psychiatry 1992;55:581–584.
123. Melo TP, Bogousslavsky J, Moulin T, et al. Thalamic ataxia. J Neurol 1992;239:331–337.
124. Mengual E, de las Heras S, Erro E, et al. Thalamic interaction between the input and the output systems of the basal ganglia. J Chem Neuroanat 1999;16: 187–200.
125. Mennemeier M, Fennell E, Valenstein E, et al. Contributions of the left intralaminar and medial thalamic nuclei to memory. Comparisons and report of a case. Arch Neurol 1992;49:1050–1058.
126. Metter EJ, Wasterlain CG, Kuhl DE, et al. FDG positron emission computed tomography in a study of aphasia. Ann Neurol 1981;10:173–183.
127. Mills RP, Swanson PD. Vertical oculomotor apraxia and memory loss. Ann Neurol 1978;4:149–153.
128. Miwa H, Hatori K, Kondo T, et al. Thalamic tremor: case reports and implications of the tremor-generating mechanism. Neurology 1996;46:75–79.
129. Mohr JP. Lacunes. Stroke 1982;13:3–11.
130. Mohr JP, Kase CS, Meckler RJ, et al. Sensorimotor stroke due to thalamocapsular ischemia. Arch Neurol 1977;34:739–741.
131. Montagna P. Fatal familial insomnia: a model disease in sleep physiopathology. Sleep Med Rev 2005;9:339– 353.
132. Morel A, Magnin M, Jeanmonod D. Multiarchitectonic and stereotactic atlas of the human thalamus [published erratum appears in J Comp Neurol 1998 February 22;391(4):545]. J Comp Neurol 1997;387:588–630.
133. Mori E, Yamadori A, Mitani Y. Left thalamic infarction and disturbance of verbal memory: a clinicoanatomical study with a new method of computed tomographic stereotaxic lesion localization. Ann Neurol 1986;20:671–676.
134. Muller A, Baumgartner RW, Rohrenbach C, et al. Persistent Kluver-Bucy syndrome after bilateral thalamic infarction. Neuropsychiatry Neuropsychol Behav Neurol 1999;12:136–139.
135. Nadeau SE, Crosson B. Subcortical aphasia. Brain Lang 1997;58:355–402; discussion 18–23.
136. Nadeau SE, Roeltgen DP, Sevush S, et al. Apraxia due to a pathologically documented thalamic infarction. Neurology 1994;44:2133–2137.
137. Neau JP, Bogousslavsky J. The syndrome of posterior choroidal artery territory infarction. Ann Neurol 1996; 39:779–788.
138. Newlin DB, Tramontana MG. Neuropsychological findings in a hyperactive adolescent with subcortical brain pathology. Clin Neuropsych 1981;2:178.
139. Noda S, Mizoguchi M, Yamamoto A. Thalamic experiential hallucinosis. J Neurol Neurosurg Psychiatry 1993;56:1224–1226.
140. Ohno T, Bando M, Nagura H, et al. Apraxic agraphia due to thalamic infarction. Neurology 2000; 54:2336–2339.
141. Ongerboer de Visser BW. Corneal reflex latency in lesions of the lower postcentral region. Neurology 1981;31:701–707.
142. Ongerboer de Visser BW, Moffie D. Effects of brain-stem and thalamic lesions on the corneal reflex: an electrophysiological and anatomical study. Brain 1979;102:595–608.
143. Ostendorf F, Liebermann D, Ploner CJ. Human thalamus contributes to perceptual stability across eye movements. Proc Natl Acad Sci U S A 2010;107: 1229–1234.
144. Papavassiliou E, Rau G, Heath S, et al. Thalamic deep brain stimulation for essential tremor: relation of lead location to outcome. Neurosurgery 2004;54: 1120–29; discussion 9–30.
145. Pepin EP, Auray-Pepin L. Selective dorsolateral frontal lobe dysfunction associated with diencephalic amnesia. Neurology 1993;43:733–741.
146. Perani D, Vallar G, Cappa S, et al. Aphasia and neglect after subcortical stroke. A clinical/cerebral perfusion correlation study. Brain 1987;110:1211– 1229.
147. Percheron G. [Arteries of the human thalamus. I. Artery and polar thalamic territory of the posterior communicating artery]. Rev Neurol (Paris) 1976;132: 297–307.
148. Percheron G. [Arteries of the human thalamus. II. Arteries and paramedian thalamic territory of the communicating basilar artery]. Rev Neurol (Paris) 1976; 132:309–324.
149. Percheron G. [Arteries of the thalamus in man. Choroidal arteries. I. Macroscopic study of individual variations. II. Systematization]. Rev Neurol (Paris) 1977; 133:533–545.
150. Peru A, Fabbro F. Thalamic amnesia following venous infarction: evidence from a single case study. Brain Cogn 1997;33:278–294.
151. Pessin MS, Adelman LS, Prager RJ, et al. “Wrong-way eyes” in supratentorial hemorrhage. Ann Neurol 1981;9:79–81.
152. Plum F, Posner JB. The diagnosis of stupor and coma, 3rd ed. Philadelphia, PA: Davis, 1980.
153. Popken GJ, Bunney WE Jr, Potkin SG, et al. Subnucleus-specific loss of neurons in medial thalamus of schizophrenics. Proc Natl Acad Sci U S A 2000;97:9276–9280.
154. Porta M, Brambilla A, Cavanna AE, et al. Thalamic deep brain stimulation for treatment-refractory Tourette syndrome: two-year outcome. Neurology 2009; 73:1375–1380.
155. Powers JM. Blepharospasm due to unilateral diencephalon infarction. Neurology 1985;35:283– 284.
156. Price DD. Psychological and neural mechanisms of the affective dimension of pain. Science 2000;288: 1769–1772.
157. Raffaele R, Tornali C, Genazzani AA, et al. Transient global amnesia and cerebral infarct: a case report. Brain Inj 1995;9:815–818.
158. Raymer AM, Moberg P, Crosson B, et al. Lexical-semantic deficits in two patients with dominant thalamic infarction. Neuropsychologia 1997;35:211–219.
159. Reynolds AF, Turner PT, Harris AB, et al. Left thalamic hemorrhage with dysphasia: a report of five cases. Brain Lang 1979;7:62–73.
160. Roiser JP, Stephan KE, den Ouden HE, et al. Adaptive and aberrant reward prediction signals in the human brain. Neuroimage 2010;50:657–664.
161. Roland PE, Nielsen VK. Vibratory thresholds in the hands: comparison of patients with suprathalamic lesions with normal subjects. Arch Neurol 1980;37: 775–779.
162. Rousseaux M, Muller P, Gahide I, et al. Disorders of smell, taste, and food intake in a patient with a dorsomedial thalamic infarct. Stroke 1996;27:2328–2330.
163. Sandson TA, Daffner KR, Carvalho PA, et al. Frontal lobe dysfunction following infarction of the left-sided medial thalamus. Arch Neurol 1991;48:1300–1303.
164. Schaltenbrand G, Bailey P. Introduction to stereotaxis with an atlas of the human brain. Stuttgart, Germany: Thieme, 1959.
165. Schmahmann JD. Vascular syndromes of the thalamus. Stroke 2003;34:2264–2278.
166. Schmahmann JD, Leifer D. Parietal pseudothalamic pain syndrome. Clinical features and anatomic correlates. Arch Neurol 1992;49:1032–1037.
167. Schnider A, Ptak R. Spontaneous confabulators fail to suppress currently irrelevant memory traces. Nat Neurosci 1999;2:677–681.
168. Schnider A, von Daniken C, Gutbrod K. The mechanisms of spontaneous and provoked confabulations. Brain 1996;119:1365–1375.
169. Schuurman PR, Bosch DA, Bossuyt PM, et al. A comparison of continuous thalamic stimulation and thalamotomy for suppression of severe tremor. N Engl J Med 2000;342:461–468.
170. Scott SK, Holmes A, Friston KJ, et al. A thalamo-prefrontal system for representation in executive response choice. Neuroreport 2000;11:1523–1527.
171. Segarra JM. Cerebral vascular disease and behaviour. I. The syndrome of the mesencephalic artery (basilar artery bifurcation). Arch Neurol 1970;22:408.
172. Sela L, Sacher Y, Serfaty C, et al. Spared and impaired olfactory abilities after thalamic lesions. J Neurosci 2009;29:12059–12069.
173. Serra Catafau J, Rubio F, Peres Serra J. Peduncular hallucinosis associated with posterior thalamic infarction. J Neurol 1992;239:89–90.
174. Sforza E, Montagna P, Tinuper P, et al. Sleep-wake cycle abnormalities in fatal familial insomnia. Evidence of the role of the thalamus in sleep regulation. Electroencephalogr Clin Neurophysiol 1995;94:398– 405.
175. Shuren JE, Jacobs DH, Heilman KM. Diencephalic temporal order amnesia. J Neurol Neurosurg Psychiatry 1997;62:163–168.
176. Siatkowski RM, Schatz NJ, Sellitti TP, et al. Do thalamic lesions really cause vertical gaze palsies? J Clin Neuroophthalmol 1993;13:190–193.
177. Solomon DH, Barohn RJ, Bazan C, et al. The thalamic ataxia syndrome. Neurology 1994;44:810–814.
178. Speedie LJ, Heilman KM. Anterograde memory deficits for visuospatial material after infarction of the right thalamus. Arch Neurol 1983;40:183–186.
179. Squire LR, Moore RY. Dorsal thalamic lesion in a noted case of human memory dysfunction. Ann Neurol 1979;6:503–506.
180. Steeves TD, Ko JH, Kideckel DM, et al. Extrastriatal dopaminergic dysfunction in Tourette syndrome. Ann Neurol 2010;67:170–181.
181. Stell R, Davis S, Carroll WM. Unilateral asterixis due to a lesion of the ventrolateral thalamus. J Neurol Neurosurg Psychiatry 1994;57:878–880.
182. Steriade M. Sleep, epilepsy and thalamic reticular inhibitory neurons. Trends Neurosci 2005;28:317–324.
183. Tatemichi TK, Steinke W, Duncan C, et al. Paramedian thalamopeduncular infarction: clinical syndromes and magnetic resonance imaging. Ann Neurol 1992;32:162–171.
184. Tatu L, Moulin T, Martin V, et al. Unilateral pure thalamic asterixis: clinical, electromyographic, and topographic patterns. Neurology 2000;54:2339–2342.
185. Taylor RB, Wennberg RA, Lozano AM, et al. Central nystagmus induced by deep-brain stimulation for epilepsy. Epilepsia 2000;41:1637–1641.
186. Temel Y, van Lankveld JJ, Boon P, et al. Deep brain stimulation of the thalamus can influence penile erection. Int J Impot Res 2004;16:91–94.
187. Tham WW, Stevenson RJ, Miller LA. The functional role of the medio dorsal thalamic nucleus in olfaction. Brain Res Rev 2009;62:109–126.
188. Theodore WH, Fisher RS. Brain stimulation for epilepsy. Lancet Neurol 2004;3:111–118.
189. Tijssen CC. Contralateral conjugate eye deviation in acute supratentorial lesions. Stroke 1994;25:1516– 1519.
190. Tinuper P, Montagna P, Medori R, et al. The thalamus participates in the regulation of the sleep-waking cycle. A clinico-pathological study in fatal familial thalamic degeneration. Electroencephalogr Clin Neurophysiol 1989;73:117–123.
191. Tuszynski MH, Petito CK. Ischemic thalamic aphasia with pathologic confirmation. Neurology 1988;38: 800–802.
192. Tyvaert L, Chassagnon S, Sadikot A, et al. Thalamic nuclei activity in idiopathic generalized epilepsy: an EEG-fMRI study. Neurology 2009;73:2018–2022.
193. Vallar G. Extrapersonal visual unilateral spatial neglect and its neuroanatomy. Neuroimage 2001;14: S52–S58.
194. Van der Werf YD, Witter MP, Groenewegen HJ. The intralaminar and midline nuclei of the thalamus. Anatomical and functional evidence for participation in processes of arousal and awareness. Brain Res Brain Res Rev 2002;39:107–140.
195. Van der Werf YD, Witter MP, Uylings HB, et al. Neuropsychology of infarctions in the thalamus: a review. Neuropsychologia 2000;38:613–627.
196. Velasco F, Velasco M. A reticulothalamic system mediating proprioceptive attention and tremor in man. Neurosurgery 1979;4:30–36.
197. Velasco M, Velasco F, Velasco AL, et al. Electrocortical and behavioral responses produced by acute electrical stimulation of the human centromedian thalamic nucleus. Electroencephalogr Clin Neurophysiol 1997;102:461–471.
198. Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff syndrome. A clinical and pathological study of 245 patients, 82 with post-mortem examinations. Philadelphia, PA: Davis, 1971:1–206.
199. Volkmann J. Deep brain stimulation for the treatment of Parkinson’s disease. J Clin Neurophysiol, 2004;21: 6–17.
200. Von Cramon DY, Hebel N, Schuri U. A contribution to the anatomical basis of thalamic amnesia. Brain 1985;108:493.
201. Vuilleumier P, Chicherio C, Assal F, et al. Functional neuroanatomical correlates of hysterical sensorimotor loss. Brain 2001;124:1077–1090.
202. Vuilleumier P, Ghika-Schmid F, Bogousslavsky J, et al. Persistent recurrence of hypomania and prosopoaffective agnosia in a patient with right thalamic infarct. Neuropsychiatry Neuropsychol Behav Neurol 1998;11:40–44.
203. Walshe TM, Davis KR, Fisher CM. Thalamic hemorrhage: a computed tomographic-clinical correlation. Neurology 1977;27:217–222.
204. Ward R, Arend I. An object-based frame of reference within the human pulvinar. Brain 2007;130:2462–2469.
205. Watson RT, Heilman KM. Thalamic neglect. Neurology 1979;29:690–694.
206. Watson RT, Valenstein E, Heilman KM. Thalamic neglect. Possible role of the medial thalamus and nucleus reticularis in behavior. Arch Neurol 1981;38: 501–506.
207. Weisbrod M. The significance of vertical gaze palsy in the paramedian thalamic artery syndrome. Neuroophthalmology 1992;12:85.
208. Westmoreland BF, Groover RV, Klass DW. Spontaneous sleep and induced arousal. A depth-electroencephalographic study. J Neurol Sci 1976;28:353–360.
209. Wilkins RH, Brody IA. The thalamic syndrome. Arch Neurol 1969;20:559.
210. Young KA, Manaye KF, Liang C, et al. Reduced number of mediodorsal and anterior thalamic neurons in schizophrenia. Biol Psychiatry 2000;47:944–953.
211. Ziegler DK, Kaufman A, Marshall HE. Abrupt memory loss associated with thalamic tumor. Arch Neurol 1977;34:545–548.
212. Zihl J, von Cramon D. The contribution of the ‘second’ visual system to directed visual attention in man. Brain 1979;102:835–856.
< div class='tao-gold-member'>




