CHAPTER 202 Cerebellar Astrocytomas
Historical Perspective
Harvey Cushing in 1931 described his experience with 76 cerebellar astrocytomas and was the first to note that they represented a group separate from other astrocytomas. He remarked on their cerebellar location, frequent cystic appearance, occurrence at a young age, and excellent prognosis after surgical resection.1 In this series, the mean age at initial evaluation was 13 years, but most of his patients had been symptomatic for 2 or more years and exhibited blindness secondary to chronic papilledema. Cushing himself suggested that improved diagnostic techniques would allow diagnosis at a younger age, and the development of computed tomography (CT) and magnetic resonance imaging (MRI) has fulfilled this prediction.
The first histologic description of these tumors was provided by Bergstrand in 1932.2 He found that many of the cells in these masses were unipolar or bipolar spongioblasts, reminiscent of cells found during the late embryonic stage of development. He suggested that these were not true neoplasms but congenital malformations. Bucy and Gustafson recognized cerebellar astrocytoma as a true neoplastic entity and first described Rosenthal’s fibers as a histologic feature.3 In the 1940s, the designation “spongioblastoma” was used to describe many of these tumors, but in 1977, the term juvenile pilocytic astrocytoma was introduced by Russell and Rubinstein.4 This term remains in the current World Health Organization classification of brain tumors.
Etiology
The cause of these tumors remains obscure. The predominance of the lesion during childhood prompted Cushing to suggest a congenital origin, and in fact, infants younger than 1 year have been reported with cerebellar astrocytoma.5 Although many patients give a history of some antecedent trauma, this is hardly surprising in a population composed of children. It is likely that the injury merely calls attention to a preexisting problem; no link between trauma or any other external etiologic factor and cerebellar astrocytoma has ever been established. There does not appear to be an increased familial incidence, and they are mainly sporadic. The incidence is equal by gender.
A germline mutation or deletion of the neurofibromatosis type 1 (NF1) gene predisposes individuals to the development of a variety of tumors, including cerebellar astrocytoma.6 A role has been postulated for deletion of the long arm of chromosome 17q or mutation in the NF1 locus in the pathogenesis of sporadic pilocytic astrocytoma, but this hypothesis has yet to be confirmed.7 Most recently, microarray technology has provided a rapid method to screen for multiple gene abnormalities in tumors. A single novel duplication in chromosome band 7q34 was identified in 20 of 28 low-grade astrocytomas.8 One known oncogene (BRAF) maps to this region.
Patient Characteristics
Incidence
Cerebellar astrocytoma accounts for between 12% and 28% of all pediatric brain tumors. Approximately half of all tumors in children arise within the infratentorial compartment, and of these, astrocytoma accounts for about a third and is equal in incidence to medulloblastoma/primitive neuroectodermal tumor.8
Age
Roughly 70% of cerebellar astrocytomas are diagnosed in children.9 Cerebellar astrocytomas may occur in adults, but the outlook is less favorable, and most likely adult tumors are a different biologic entity, similar to astrocytomas in other locations. In children, the age at diagnosis has been steadily declining as imaging techniques improve and become more readily available. In 1971, the average age at admission to the hospital was 8.9 years.10 More recent series published in the era of modern imaging report the average age to be 7 years.11 Cerebellar astrocytoma is rare in infants. A midline cerebellar tumor in a young child or infant is more likely to be a medulloblastoma or ependymoma.
Clinical Findings
The typical history is characteristic and indistinguishable from that of other extrinsic posterior fossa masses apart from the duration of symptoms. Usually, the child has nonspecific symptoms of raised intracranial pressure arising from obstructive hydrocephalus. The symptoms are frequently insidious and, in retrospect, may have been present intermittently for years. The slow growth of these tumors allows gradual displacement of the adjacent cerebellum and brainstem, and alarming symptoms usually await the development of massive tumors and hydrocephalus. In small children, the cranium can expand to accommodate the increased intracranial volume with little increase in pressure. The early symptoms that do occur are often nonspecific and frequently attributed to viral illness, migraine, gastrointestinal disease, or psychiatric problems. The duration of symptoms before definitive diagnosis has fallen from 18.7 months reported by Ilgren and Stiller9 to 5.8 months in a more recent report.12 A long history of headache or vomiting in the context of a posterior fossa mass suggests the diagnosis of astrocytoma, whereas a history of just a few days or weeks makes a more rapidly growing tumor such as ependymoma or medulloblastoma more probable. The abundance of CT and MRI scanners in the developed world has resulted in an increased incidence of asymptomatic cerebellar astrocytomas found as incidental lesions on imaging performed for unrelated indications such as trauma.
Headache is the most common symptom in children with posterior fossa tumors and occurs in virtually 100% in some series.10 Nonspecific headache is common in children, but the headache of a posterior fossa tumor is characteristic. It is more often frontal than suboccipital at first. When the headache becomes localized to the suboccipital region, it is often described along with neck pain or stiffness and suggests chronic tonsillar herniation. Headaches occurring only in the morning and subsiding with activity are particularly suggestive. The combination of hypoventilation associated with sleep and increased intracranial pressure with recumbence provokes the headache on awakening, and the pain may awaken the child from sleep. Coughing, sneezing, or straining at stool may cause headache, and some children may become constipated to avoid discomfort.
Other signs and symptoms may include macrocephaly, personality change, torticollis, and dizziness. Diplopia from sixth nerve palsy may accompany hydrocephalus. Other cranial neuropathies suggest brainstem involvement. Tumors in the vermis tend to come to medical attention at a younger age, and patients complain mainly of headache and vomiting. Laterally placed tumors in the cerebellar hemisphere are more likely to occur in older patients and are more likely to be associated with dysmetria and tremor.5
Imaging Features
Cushing described the classic appearance of a cyst associated with a discrete mural nodule found at surgery. With modern imaging this picture has changed, and this classic appearance is present in less than 50% of cases.1,13 Cerebellar astrocytomas appear on CT or MRI in one of several predominant forms:
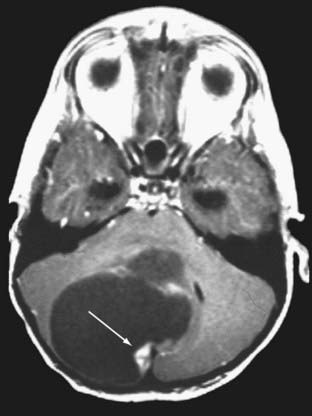
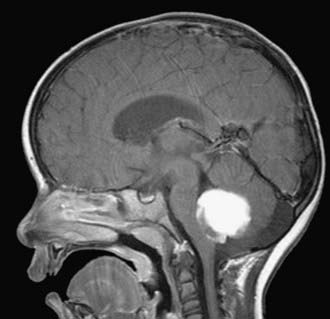
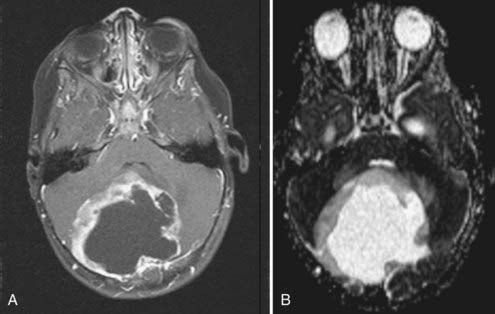
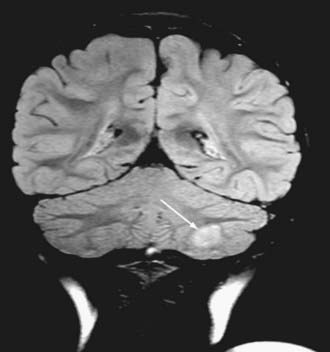
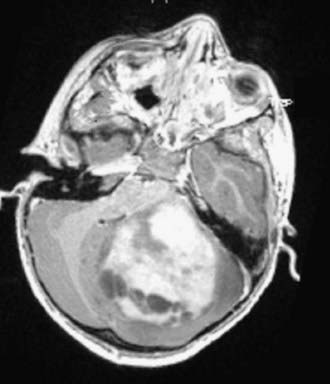
It is estimated that tumors are strictly confined to the cerebellum in 76% and involve the brainstem or cerebellar peduncle (“transitional forms”) in 24%.11
Unusual radiographic findings have been reported. Leptomeningeal dissemination at diagnosis is extremely rare but has been reported at the time of recurrence.19 Some degree of radiographic tonsillar herniation is common, particularly with large tumors, and cervical hydrosyringomyelia has been reported as well, presumably from interference with CSF flow at the foramen magnum.19 A case of a densely calcified cerebellar astrocytoma in an infant has been published as well.20
In summary, the appearance of a cerebellar astrocytoma in a child is sufficiently characteristic that the diagnosis is usually made preoperatively. A combination of radiographic and clinical findings makes the diagnosis even more accurate, and neural network programs have exhibited accuracy as high as 95% in predicting tumor type with pediatric posterior fossa tumors.21
Surgical and Perioperative Management
The primary goal of surgery for cerebellar astrocytoma is total resection of the mass. If contrast-enhanced MRI performed 6 months after surgery, when the postoperative changes have resolved, shows no residual tumor, the patient is very likely cured.22 This is possible in about 88% of cases.11 Total resection is usually straightforward in patients with cystic tumors and a laterally placed mural nodule, but it may be limited by involvement of the brainstem or cerebellar peduncle.
Severe symptoms are generally the result of obstructive hydrocephalus rather than the mass itself, and considerable debate has centered on the management of hydrocephalus. Options include steroids followed by tumor removal,23 external ventricular drainage,24 placement of a shunt before removal of the tumor,25 and preoperative endoscopic third ventriculostomy.26,27 Advocates of preoperative CSF diversion point out that with this strategy, time is afforded to prepare the patient and family for the definitive procedure, perform diagnostic tests, and provide a safer surgical procedure once the increased intracranial pressure has been relieved. Others point out the potential disadvantages: (1) only about 20% of patients with cerebellar astrocytomas ultimately require shunts postoperatively,28 and routine placement of shunts condemns the entire group to the possibility of shunt-related complications; (2) a preoperative shunt delays definitive treatment; (3) shunts serve as a potential route for dissemination should the tumor prove to be malignant; and (4) upward transtentorial herniation has been reported after acute decompression of the supratentorial compartment when the posterior fossa mass is still present.25,29 Endoscopic third ventriculostomy carries a rate of severe complications as high as 9% and is unlikely to relieve any component of hydrocephalus caused by absorptive failure after tumor removal.26
McLaurin concluded that these risks and benefits probably balance each other out and that either approach is acceptable.30 Most neurosurgeons prefer to remove the tumor, place a ventriculostomy during the surgery, and insert a shunt in the postoperative period if needed. Performance of a ventriculostomy at the bedside may be required as an emergency but should probably be followed by prompt tumor removal.
For most posterior fossa tumors in children, the prone position is recommended. The risk for air emboli with the sitting position is largely eliminated, and the surgeon’s arms become less fatigued. The method of head support depends on the age of the patient. Infants and very young children are probably best supported with a padded horseshoe headrest, and care must be taken to avoid pressure on the eyes, forehead, and malar eminences. Older children (≥2 years) and those with high vermian lesions in whom head flexion is desirable are best managed with pin fixation. The Sugita headrest is useful for younger children because multiple fixation points are used and thus require less force. The standard Mayfield device may be used in older children (Fig. 202-6). Patients with long-standing hydrocephalus may have very thin skulls, so care should be taken when adjusting the force of the pins. Severe flexion may result in kinking of the endotracheal tube, high inflating airway pressure, and venous bleeding. Nasotracheal intubation and communication with the anesthesiologist are helpful.
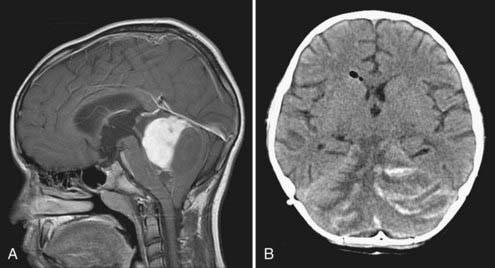
Laterally placed tumors with a cyst and a mural nodule may be approached by a transverse corticectomy through the cerebellar folia. Midline tumors usually require at least some splitting of the cerebellar vermis. A cerebellomedullary fissure approach has been described and allows relatively small inferior tumors to be removed without splitting the vermis, but it is unclear whether this avoids mutism or other potential problems.31 Tumors with a significant solid portion in the superior vermis may pose particular problems. They may be approached by splitting the superior vermis or by going through a supracerebellar infratentorial corridor as done for pineal region tumors. This approach involves sacrificing at least some of the bridging veins to the sinuses of the tentorium. If these veins are stretched, there is risk of tearing them from the straight sinus, which can result in profuse bleeding that is difficult to control. Sacrifice of these veins is usually well tolerated, but in some individuals unpredictable venous infarction, swelling, and hemorrhage may occur (Fig. 202-7).32






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


