CHAPTER 220 Cervical Spine Disorders in Children
The true prevalence of congenital abnormalities of the cervical spine is unknown, although the incidence is probably underrepresented because many are asymptomatic. Some authors have reported that congenital anomalies of the cervical spine in children with Klippel-Feil syndrome (KFS), a relatively common congenital spine disorder, occur in 1 in 40,000 to 42,000 births.1–3 Population-based studies to estimate the incidence and prevalence of congenital cervical spine abnormalities are not available. Although some developmental abnormalities are isolated sporadic cases, many are found as part of a multisystem or syndromic anomaly (Table 220-1).4
Working knowledge of the anatomy and embryology of the pediatric cervical spine is crucial to properly identify congenital abnormalities, differentiate congenital from traumatic lesions, and approach the region at surgery. We refer the reader to several publications that describe pediatric cervical spine anatomy and embryology in detail.5–26
Specific Conditions
Down Syndrome
Down syndrome (trisomy 21) is the most common inherited chromosomal disorder, with an estimated prevalence of 1 in 700 live births. Occipitoatlantal subluxation and atlantoaxial subluxation are the two most common cervical spine problems seen in patients with Down syndrome. Patients with symptomatic atlantoaxial or occipitocervical instability may suffer from neck pain, torticollis, myeloradiculopathy, spasticity, hyperreflexia, or clonus.27,28
Occipitocervical instability is present to some degree in 40% to 50% of children with Down syndrome. To date, no studies have looked at the natural history of this problem. The normal occipitoatlantal joint is composed of reniform-shaped occipital condyles seated on cup-shaped articular facets supported by the capsular ligaments.29 Analysis of the occipitocervical junction in children with Down syndrome demonstrated that the superior articular surface of C1 and the occipital condyle are flattened in these patients.30 Surgical treatment is generally recommended in patients who have more than 10 mm of subluxation at the occipitoatlantal joint, although that decision must be made on an individual basis because of the low incidence of spinal cord injury in otherwise asymptomatic patients.29–31
Atlantoaxial subluxation is related to either the excessive ligamentous laxity or the osseous abnormalities in patients with Down syndrome. Radiographic atlantoaxial instability, as determined by the atlantodental interval and measurements of spinal canal diameter, is present in 10% to 30% of patients with Down syndrome,29,32–35 although only 1% of such patients have symptomatic C1-2 instability.27,28 One study of pediatric patients with Down syndrome and symptomatic atlantoaxial instability found that more than 80% of patients had os odontoideum.28
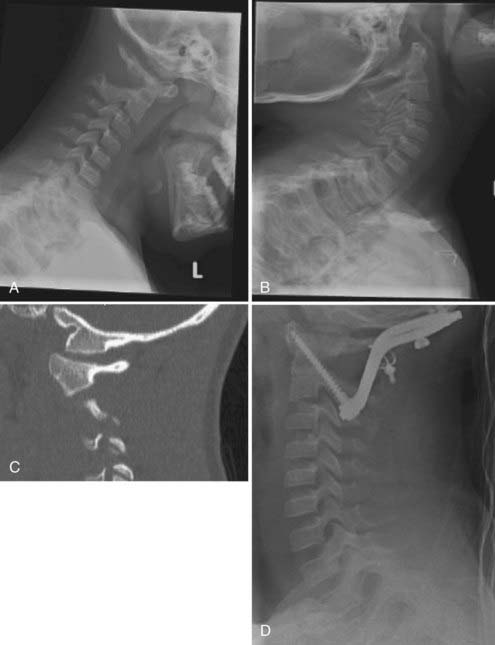
Suggestions have been made to screen children with Down syndrome for instability (Fig. 220-1).29,34,36 Cervical radiographs, including flexion and extension views, should be performed initially at 3 years of age to assess for excessive motion. If radiographs are normal, they may be repeated every 5 years or at the time of consideration for participation in the Special Olympics.29,34,36 For patients with abnormal results on screening studies (occipitocervical subluxation >10 mm, atlantodental interval >4.5 mm, or neural canal <12 mm), careful yearly follow-up is indicated. Surgical correction may be required if neurological signs and symptoms are present, if magnetic resonance imaging (MRI) shows spinal cord injury, if subluxation is progressive over time, or if an os odontoideum is present.29
Klippel-Feil Syndrome
First described in 1912,37 the now-classic triad of a short neck, low hairline, and limited neck mobility is known as Klippel-Feil syndrome, although less than 50% of patients with congenital fusion of the cervical spine have all three signs. Patients without all three signs are considered to have Klippel-Feil variant.1
The most common clinical finding in patients with KFS is a limited range of neck motion, particularly with lateral bending. Patients with extensive neck fusion and fusion of the craniovertebral junction are seen at an earlier age than those with subaxial fusion.4 Patients with C1-2 fusion are often first evaluated for pain in childhood, whereas those with lower cervical fusion are seen in the second or third decade of life or later when symptomatic junctional degeneration develops. Torticollis or neck webbing is present in just 20% of patients with KFS and is not specific to any particular cervical spine anomaly.38,39 Neurological abnormalities seen in patients with KFS include brainstem dysfunction, myeloradiculopathy, and headaches.
Among several classification schemes for KFS developed over the years,39–41 the most recent, presented by Tracy and colleagues, separates KFS into four classes and incorporates the mode of inheritance (Table 220-2).40
TABLE 220-2 Klippel-Feil Syndrome Types
| TYPE | INHERITANCE PATTERN | CHARACTERISTICS |
|---|---|---|
| KF1 | Autosomal recessive | Fusion at C1, with or without caudal fusion; associated with other anomalies |
| KF2 | Autosomal dominant | 100% penetrance of C2-3 fusion |
| KF3 | Autosomal dominant or recessive | Congenital fusions caudal to C2-3 |
| KF4 (Wildervanck’s syndrome) | X-linked dominantly inherited pattern; hemizygous lethal | Congenital cervical fusion, hearing loss, Duane’s anomaly (congenital disorder of eye movement) |
Achondroplasia
Achondroplasia, the most common form of dwarfism, is characterized by disproportionate shortening of the proximal ends of limbs relative to the trunk. It occurs in 1 in 26,000 to 28,000 births, with an estimated incidence of 0.03% to 0.05% of all live births.42–44 It is inherited in autosomal dominant fashion, with 80% of cases attributed to spontaneous point mutations in the fibroblast growth factor receptor 3 gene located on chromosome 4.42,44 The cause of achondroplasia is a decrease in the rate of endochondral bone formation with normal formation of membranous bone.
The predominant spinal condition in achondroplasia is compression, which can occur at the foramen magnum, subaxial cervical spine, or thoracolumbar spine (Fig. 220-2). The clinical features of patients with cervicomedullary compression secondary to stenosis of the foramen magnum include bulbar findings, myeloradiculopathy, hydrocephalus, and respiratory disorders. Symptoms of cervicomedullary compression in infants include excessive hypotonia, poor head control, feeding or sleep abnormalities, and apnea. The respiratory disturbances may have a multifactorial cause. Neurological findings may be difficult to detect in young patients because many children with achondroplasia are normally hypotonic during early infancy, although they often exhibit developmental delay in achieving motor milestones.45 A small percentage of patients with stenosis of the foramen magnum have symptoms of cervicomedullary compression.45,46 In a prospective evaluation of 53 infants with achondroplasia, more than 70% had foramen magnum stenosis, but less than 10% demonstrated clinical symptoms of cervicomedullary compression.46
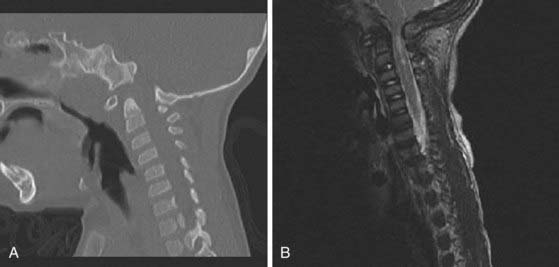
Sudden death as a catastrophic consequence of cervicomedullary compression has been reported in infants with achondroplasia.46,47 Hecht and colleagues reported a 7.5% risk for sudden death within the first year of life and a 2.5% risk for sudden death between 1 and 4 years of age in children with achondroplasia.47
Goldenhar’s Syndrome
Spinal abnormalities associated with GS include vertebral hypoplasia, failure of segmentation, and failure of vertebral formation. Segmentation defects are more common in the cervical spine, whereas vertebral formation abnormalities more frequently occur in the thoracolumbar spine. In one series, 60% of children with GS had vertebral anomalies,48 including block vertebrae (most often involving fusion of C3 and C4), unilateral thoracolumbar hemivertebrae, spina bifida occulta, butterfly vertebrae, and sacral agenesis. Many patients had abnormalities at multiple spinal levels.
Upper cervical spine anomalies may also be seen in association with GS. One group reported a 12% incidence of vertebral segmentation anomalies and occipitalization of the atlas.48 Odontoid hypoplasia with atlantoaxial instability has also been reported.49
Spondyloepiphysial Dysplasia
The most common spinal abnormalities in children with SED are odontoid hypoplasia and ligamentous laxity. Spinal cord compression from atlantoaxial instability causes myelopathy in as many as 35% of children with SED. The myelopathy usually develops gradually and is manifested as delayed motor development, slowly progressive weakness, spasticity, sleep apnea, and other respiratory abnormalities. Atlantoaxial instability has been found to progress with age in SED. Those with spinal canal diameters smaller than 10 mm at the level of the axis are at significantly increased risk for spinal cord compression.50,51
Morquio’s Syndrome
Mucopolysaccharidosis (MPS) type IV, or Morquio’s syndrome, is an autosomal recessive lysosomal storage disease that results from an inability to metabolize keratan sulfate, a glycosaminoglycan found predominantly in cartilage and the cornea.52,53 Patients often appear normal at birth and have normal growth and development for the first 2 years of life until the underlying skeletal abnormalities become clinically apparent between 2 and 6 years of age. The incidence of MPS IV is 1 in 40,000 live births. Many patients survive to early adulthood, at which time they may succumb to the cardiopulmonary and neurological complications of the disorder.
The most common spinal abnormality is atlantoaxial subluxation with spinal cord compression.54 Atlantoaxial subluxation has been identified in 42% to 90% of patients with Morquio’s syndrome.55 Odontoid dysplasia, which may be manifested as hypoplasia, aplasia, or os odontoideum, is common in MPS IV.
Osteogenesis Imperfecta
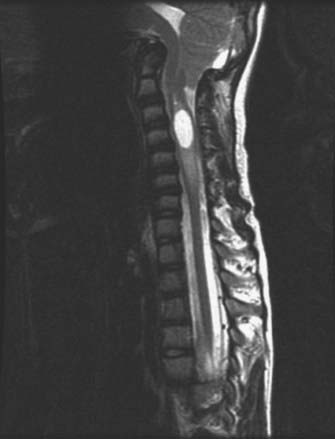
Osteogenesis imperfecta (OI) is a hereditary disorder caused by a defect in collagen production. The disease is characterized by osteopenia, fragile bones susceptible to fracture, short stature, and progressive skeletal deformity.56 The most commonly used classification system for OI is the Sillence classification system.57,58 Type I is mild with no long bone deformities, type II is lethal in the perinatal period with in utero fractures, type III is the most severe form in children who survive the perinatal period, and type IV is an undefined type with moderate bone deformities and variable short stature. Abnormalities at the craniovertebral junction are most commonly seen with type IV OI, with a prevalence ranging from rare to 25%.57 Osteoporotic bones resulting in repetitive microfracture cause infolding of the occipital condyles with elevation of the clivus and posterior cranial fossa. This leads to upward migration of the cervical spine into the foramen magnum and symptomatic compression of the brainstem and lower cranial nerves (Fig. 220-3).59
Larsen’s Syndrome
Larsen’s syndrome is a rare congenital disorder of connective tissue.60–69 The cervical spine is more commonly affected than the thoracic or lumbar spine. The most characteristic abnormalities include hypoplastic or flattened vertebral bodies, dysraphism, hemivertebrae, and wedged vertebrae.63,64 Midcervical subluxations and kyphosis have been described in 12% of patients with Larsen’s syndrome.64,70 With progression of the curve and increased instability, progressive myelopathy, segmental weakness, and even death may ensue as a result of respiratory insufficiency.61–6365 Cervical kyphosis associated with anterior-posterior disconnection syndrome has also been described.66,67,71






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


