Chapter 64 Channelopathies
Episodic and Electrical Disorders of the Nervous System

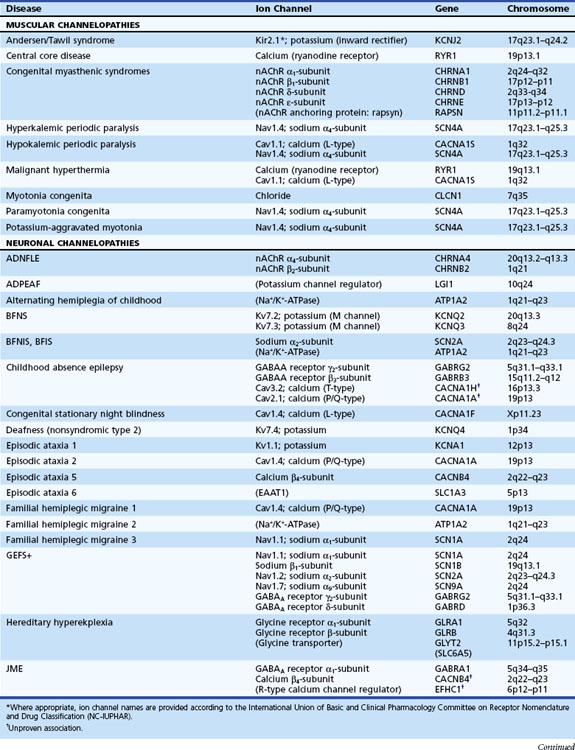
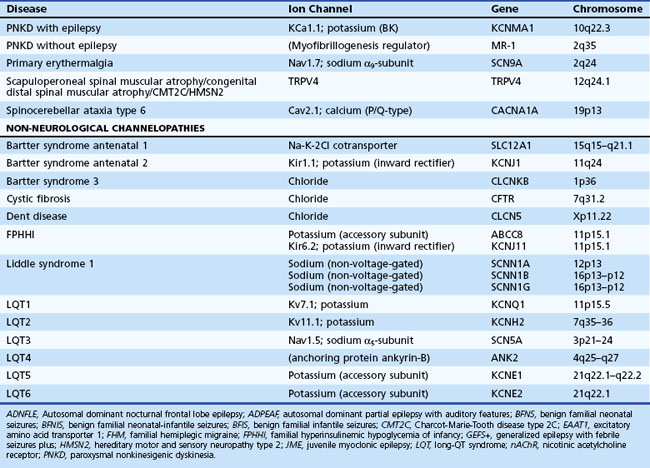
Channelopathies are disorders caused by ion channel dysfunction. Because of the great diversity of ion channel proteins and their expression in different tissues, channelopathies comprise a wide variety of clinical diseases (Table 64.1), the discovery of which helps elucidate how ion channels function in both illness and health. The periodic paralyses—the first group of ion channel disorders characterized at a molecular level—defined the field of channelopathies, which now encompasses diseases not only in muscle but also in the kidney (Bartter syndrome), epithelium (cystic fibrosis), and heart (long QT syndrome) as well as neurons. Because muscles and neurons are electrical organs, it is not surprising that most channelopathies are associated with neurological disease. Despite significant heterogeneity, a pervasive feature of neurological channelopathies is a paroxysmal phenotype. After a brief introduction to ion channels, this chapter describes disorders caused by congenital and acquired dysfunction of ion channels expressed in skeletal muscle and neurons.
Ion Channels
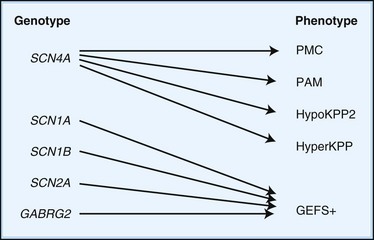
Depending on the location within the channel, mutations could alter voltage-dependent activation, ion selectivity, or time and voltage dependence of inactivation. Thus, two different mutations within the same gene can result in dramatically different physiological defects. For example, a mutation that prevents or slows inactivation could lead to a persistent ionic current. Conversely, a mutation elsewhere in the same gene that prevents activation will decrease ionic current. Phenotypic heterogeneity describes how different mutations in a single gene cause distinct phenotypes. For instance, mutations in the skeletal muscle voltage-dependent sodium channel can result in hyperkalemic periodic paralysis, hypokalemic periodic paralysis, potassium-aggravated myotonia, or paramyotonia congenita (see Table 64.1 and Fig. 64.1). In contrast, genetic heterogeneity occurs when a consistent clinical syndrome results from a variety of underlying mutations.
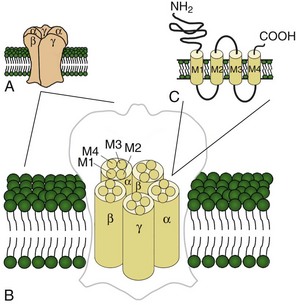
Voltage-gated potassium channels (VGKC) consist of four homologous α-subunits that combine to create a complete channel. Like the other channels described later, humans possess many distinct VGKC genes, and the resulting channels exhibit specialized properties and display rich tissue-type and cellular-compartment specificity. Each α-subunit contains six transmembrane segments (S1 to S6) linked by extracellular and intracellular loops (Fig. 64.2). The S5-S6 loop penetrates deep into the central part of the channel and lines the pore. The S4 segment contains positively charged amino acids and acts as the voltage sensor. These channels serve many functions, most notably to establish the resting membrane potential and to repolarize cells following an action potential. A unique class of potassium channel, the inwardly rectifying potassium channel, is homologous to the S5 to S6 segment of the VGKC. Because the voltage-sensing S4 domain is absent, voltage dependence results from a voltage-dependent blockade by magnesium and polyamines.
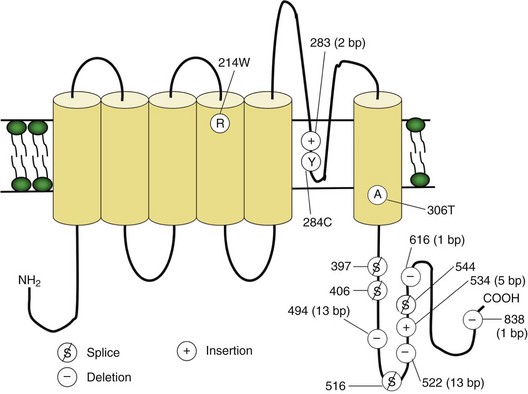
Voltage-gated sodium and calcium channels are highly homologous and share homology with VGKCs, from which they evolved. The α-subunits contain four highly homologous domains in tandem within a single transcript (DI–DIV) (Fig. 64.3). Each domain resembles a VGKC α-subunit, with six transmembrane segments as described earlier. Sodium and calcium channels differ in several regards, despite their many similarities. The amino acid sequence forming the selectivity filter and the modulatory auxiliary subunits are different. The sodium channel is composed of an α- and a β-subunit, and the calcium channel is composed of a pore-forming α1-subunit, an intracellular β-subunit, a membrane-spanning γ-subunit, and a membrane-anchoring α2δ-subunit. Sodium channels mediate fast depolarization and underlie the action potential, whereas voltage-gated calcium channels (VGCCs) mediate neurotransmitter release and allow the calcium influx that leads to second messenger effects.
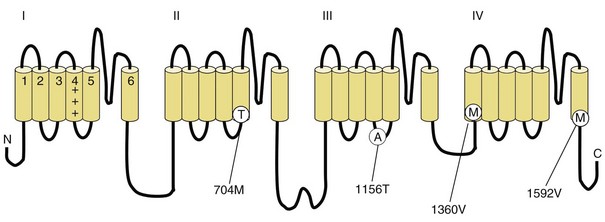
Fig. 64.3 Diagram of the voltage-gated sodium channel and the four most common mutations causing hyperkalemic periodic paralysis. The α-subunit consists of four highly homologous domains (I to IV), each containing six transmembrane segments (S1 to S6). When inserted into the membrane, the four domains fold so as to encircle a central ion-selective pore lined by the S5/S6 loop. Analogous to the potassium channel (see Fig. 64.2), the S4 segments contain positively charged residues conferring voltage dependence on the channel. Auxiliary β-subunits are not shown.
Ligand-gated ion channels activate on binding with their respective agonists. Among the diverse array of ligand-gated channels, GABAA, glycine, and nicotinic acetylcholine receptors (nAChRs) possess the only known disease-causing mutations. Although distinguished by their ligand binding and ion permeability, channels gated by GABA, glycine, and acetylcholine share several structural similarities. Five intrinsic membrane subunits assemble to form hetero- or homopentamers. Each subunit contains four transmembrane domains (M1 to M4), the second of which lines the pore and determines ionic selectivity (Fig. 64.4). Subunits contributing to nAChRs at the neuromuscular junction differ from those expressed in the central nervous system, explaining why mutation of one gene may cause seizures without affecting neuromuscular transmission, or vice versa. Binding of acetylcholine opens the channel, which conducts monovalent cations (Na+ and K+) with little or no selectivity, and some are additionally permeable to calcium. Channel activation results in membrane depolarization and excitation of the postsynaptic neuron or muscle fiber.
Genetic Disorders of Muscular Ion Channels
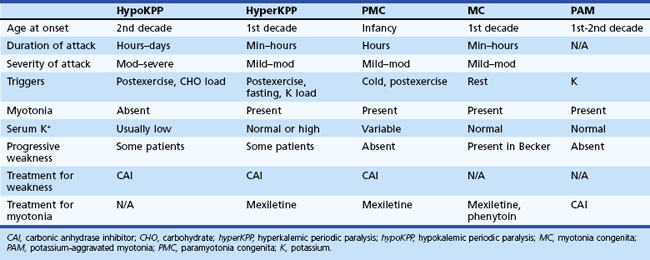
The periodic paralyses and nondystrophic myotonias encompass several skeletal muscle disorders inherited as autosomal dominant traits. These disorders include hypokalemic periodic paralysis (hypoKPP), hyperkalemic periodic paralysis (hyperKPP), paramyotonia congenita, myotonia congenita, potassium-aggravated myotonia, and Andersen-Tawil syndrome. General features include episodic weakness or stiffness, interictal return to an asymptomatic state, and responsiveness to carbonic anhydrase inhibition (see Table 64.3).
Hypokalemic Periodic Paralysis
Pathophysiology
In up to 70% of cases, the responsible mutation has been linked to a gene encoding a VGCC on chromosome 1q (Venance et al., 2006). The gene, CACNA1S, encodes the α1-subunit of the dihydropyridine-sensitive L-type VGCC found in skeletal muscle. This channel functions as the voltage sensor of the ryanodine receptor and plays an important role in excitation-contraction coupling in skeletal muscle. Four mutations in the S4 segments are responsible for voltage sensitivity. Two mutations, one involving arginine-to-histidine substitutions within the highly conserved S4 segments of DII and DIV (Arg-528-His and Arg-1239-His) account for most cases. The others involve arginine-to-glycine substitutions at the same locations.
Some 10% to 20% of families with hypoKPP have mutations in the gene encoding the α-subunit of the skeletal muscle voltage-gated sodium channel (SCN4A) on chromosome 17q. This is the same channel implicated in hyperKPP and other disorders described later (see Fig. 64.1). Evidence suggests that this sodium channel–associated syndrome is phenotypically different from the more common CACNA1S form. A proposed separate clinical entity, hypoKPP2, may be distinguishable from hypoKPP associated with CACNA1S by the presence of myalgias following paralytic attacks, and the presence of tubular aggregates instead of vacuoles in the muscle biopsy. In some patients, acetazolamide worsens symptoms (Bendahhou et al., 2001; Sternberg et al., 2001). In a large retrospective series, hypoKPP2 was associated with an older age of onset and shorter duration of attacks than classical hypoKPP (Miller et al., 2004).
Whether involving SCN4A or CACNA1S, virtually all mutations causing hypoKPP involve an S4 voltage-sensor domain. In the case of the sodium channel, these mutations allow a leak current to pass through the “gating pore” at resting membrane potentials, bypassing the central channel pore and leading to inappropriate muscle fiber depolarization and consequent channel inactivation and action potential failure (Sokolov et al., 2007). Speculation exists that this phenomenon may also occur in mutated VGCCs.
Diagnosis
An accurate medical history is essential for the diagnosis because observation of attacks is unusual, and patients are often normal between attacks. Characteristic features of hypoKPP that distinguish it from hyperKPP are that paralytic attacks are less frequent, longer lasting, precipitated by a carbohydrate load, and often begin during sleep (Table 64.2). Potassium concentrations are usually low during an attack, although concentrations less than 2 mM should suggest a secondary form of periodic paralysis. Electrocardiogram (ECG) changes such as increased PR and QT intervals, T-wave flattening, and prominent U waves suggest an underlying hypokalemia. Provocative testing can be dangerous and is not routine. Test performance requires a hospitalized setting with continuous cardiac monitoring and should be performed only in patients without cardiac or renal disease. After giving an oral glucose load (2-5 mg/kg up to a maximum of 100 g) without or, less commonly, with subcutaneous insulin (0.1 U/kg), perform serial examinations of strength while monitoring serum glucose and potassium concentrations. Other diagnostic tests include electromyography (EMG), which may show decreased compound muscle action potential amplitudes during attacks compared with interictal values. Muscle histology reveals nonspecific myopathic changes of tubular aggregates or vacuoles within fibers. Genetic testing should render muscle biopsy and provocative testing obsolete for diagnosis.
Thyrotoxic periodic paralysis may be clinically indistinguishable from hypoKPP, except that it is not familial and serum potassium levels are often lower than in familial hypoKPP (<2.5). Some cases may be associated with a mutation in KCNJ18, the gene encoding a novel inwardly rectifying potassium channel (Ryan et al., 2010). All patients with hypoKPP require screening for hyperthyroidism, as the treatment—correction of the thyroid disorder—differs from that outlined later for idiopathic hypoKPP. Exclude other secondary forms of hypokalemic paralysis when serum potassium concentrations remain low between attacks. Renal, adrenal, and gastrointestinal causes of persistent hypokalemia are common, and thiazide diuretic use or licorice (glycyrrhizic acid) intoxication are considerations.
Treatment
An effective holistic approach to treatment includes lifestyle modifications and acute and chronic pharmacological intervention. Dietary modification to avoid high carbohydrate loads and refraining from excessive exertion helps prevent attacks. Oral potassium (5-10 g load) reverses paralysis during an acute attack. Prophylactic use of acetazolamide (Table 64.3) decreases the frequency and severity of attacks. Dichlorphenamide is another carbonic anhydrase inhibitor that effectively prevents attacks, as demonstrated in a randomized clinical trial (Tawil et al., 2000) where the average dose was 100 mg daily. More potent than acetazolamide, dichlorphenamide may be useful when efficacy of the former begins to fail. Many believe, without supporting evidence, that reducing the frequency of paralytic attacks provides protection against the development of myopathy. As insight into molecular pathophysiology expands, new treatment possibilities—and a new understanding of old treatments—seem likely (Matthews and Hanna, 2010).
| Use | Prophylactic agent for some channelopathies (see text) |
| Mechanism | Inhibits carbonic anhydrase |
| Dosing | Adults: start 125 mg daily, titrating as needed up to a maximum daily dose of 1000-1500 mg, divided bid–qid. An extended release formulation is available. Children: consult a pharmacist. |
| Side effects | Taste changes (especially for carbonated drinks), fatigue, paresthesias, metabolic acidosis, blurred vision, myelosuppression, nephrolithiasis, etc. (Increased dietary citrate might be recommended to compensate for decreased urinary citrate observed during acetazolamide therapy.) |
| Monitoring | Check electrolytes, BUN, creatinine, and CBC at baseline and periodically throughout therapy. |
| Metabolism | None; excreted unchanged by kidneys |
Please note that this table is for brief informational purposes only. Prescribing physicians should consult a pharmacist or an appropriate reference for complete and updated information.
bid, Twice daily; BUN, blood urea nitrogen; CBC, complete blood cell count; qid, 4 times daily.
Hyperkalemic Periodic Paralysis
Clinical
Characteristic of this disorder is episodic weakness precipitated by hyperkalemia. Although the weakness is generally milder than in hypoKPP, it can be sufficiently severe in hyperKPP to cause flaccid quadriparesis. As in hypoKPP, respiratory and ocular muscles are unaffected and consciousness is preserved. Frequency of attacks varies from several per day to several per year. Attacks are usually brief, lasting 15 to 60 minutes, but may last up to days. Unlike hypoKPP, myotonia is present between attacks. Onset is usually in infancy or childhood, and characteristic attacks occur by adolescence. Triggers include rest after vigorous exercise, foods high in potassium, stress, and fatigue. Despite its name, hyperKPP is often associated with a normal serum potassium concentration during an attack (see following Diagnosis section). Most patients experience a subacute onset, and some describe paresthesias or a sensation of muscle tension prior to attacks. In these situations, mild exercise may abort or lessen the severity of the attack. Mild weakness may persist afterward, and the later development of a progressive myopathy is common.
Pathophysiology
HyperKPP is as an autosomal dominant disorder, with some sporadic cases. The disorder links to SCN4A, the same gene responsible for a minority of hypoKPP cases. Among several identified missense mutations, four account for about two-thirds of cases (see Fig. 64.1). Functional expression of naturally occurring mutations demonstrated a decrease in the voltage threshold of channel activation or abnormally prolonged channel opening or both (Bendahhou et al., 2002; Hayward et al., 1999), effectively increasing the depolarizing inward current. If sustained long enough, this would lead to inactivation of the sodium channels, transitory cellular inexcitability, and weakness.
Diagnosis
Despite advances in defining the underlying genetic mutations, a thorough medical and family history and physical examination remain the best diagnostic tools. Furthermore, genetic testing is not generally available, and the high number of causative sodium channel mutations makes widespread use of genetic testing unlikely in the near future. Serum potassium is normal between attacks and even during many attacks. Unlike hypoKPP, potassium administration may precipitate an attack. In the absence of provocative testing, the basis for diagnosis is the clinical presentation. Myotonia is present in many patients between attacks, either spontaneously or after muscle percussion, and failure to produce myotonia discriminates hypoKPP from hyperKPP (see Table 64.2). Take care not to confuse subjective muscle stiffness with objective changes. Peaked T waves on ECG suggest hyperkalemia and are an aid to diagnosis. As in hypoKPP, serum creatine kinase concentrations may be normal or elevated. Electrodiagnostic studies are useful for demonstrating subclinical myotonic discharges, not seen in hypoKPP. Nonspecific findings such as fibrillation potentials and small polyphasic motor unit potentials occur during late stages of disease.
Treatment
The goal of therapy is to abort the acute attacks and prevent future attacks. Acute attacks are often sufficiently brief and mild so as not to require acute intervention. In more severe attacks, aim treatment at lowering extracellular potassium levels. Mild exercise or eating a high sugar load (juice or a candy bar) may suffice, as insulin drives extracellular potassium into cells. Thiazide diuretics and inhaled β-adrenergic agonists are similarly helpful, and intravenous calcium gluconate may be useful when weakness is very severe. To prevent attacks, a diet low in potassium and high in carbohydrates may obviate the need for prophylactic drug therapy. Oral dichlorphenamide was useful for prophylaxis in one randomized controlled trial (Tawil et al., 2000). Acetazolamide (see Table 64.3) and thiazide diuretics are useful as well. Successful prophylaxis may decrease the later onset of myopathy, although direct proof of this hypothesis is lacking. Finally, myotonic symptoms are troublesome in some patients; based on the underlying pathophysiology, sodium channel blockers would seem an effective therapy, and mexiletine is commonly used for this purpose.
Paramyotonia Congenita
Treatment
Symptoms are generally mild and infrequent. Direct treatment, when required, at either myotonia or weakness or both. Sodium channel blockers such as mexiletine are sometimes effective in reducing the frequency and severity of myotonia. Patients with weakness often respond to agents used to treat hyperKPP (e.g., thiazides, acetazolamide). A single case report suggests the possible use of pyridostigmine (Khadilkar et al., 2010). Cold avoidance reduces the frequency of attacks.
Myotonia Congenita
Clinical
Inheritance of myotonia congenita (MC) is either as an autosomal dominant (Thomsen disease) or recessive (Becker myotonia) trait. The main feature is myotonia or delayed muscle relaxation after contraction. Forceful movement abruptly initiated after several minutes of rest causes the most pronounced myotonic stiffness. The myotonia of MC displays a warm-up phenomenon in which the myotonia decreases or vanishes completely when repeating the same movement several times, in contrast to the myotonia seen in patients with PMC. The onset of Thomsen myotonia is often within the first decade, whereas the onset of Becker myotonia is generally at 10 to 14 years of age. Although myotonia can affect all skeletal muscles, it is especially prominent in the legs, where it is occasionally severe enough to impede a patient’s ability to walk or run. In rare cases, sudden noise causes sufficient generalized stiffness to make the patient fall to the ground and remain rigid for several seconds. The recessive and dominant forms share many similarities, but some clinical features help distinguish the two. In general, patients with recessive disease experience transitory bouts of weakness after periods of disuse and may develop progressive myopathy (Kornblum et al., 2010); in addition, muscle hypertrophy and disease severity are greater than in the dominant form. Becker myotonia is more common than Thomsen disease.
Pathophysiology
Electrical instability of the sarcolemma leads to muscle stiffness by causing repetitive electric discharges of affected muscle fibers. Early in vivo studies in myotonic goats revealed greatly diminished sarcolemmal chloride conductance in affected muscle fibers. This causes a depolarization of the sarcolemmal membrane and muscle hyperexcitability. Genetic linkage analysis for both recessive and dominant forms of MC pointed to a locus on chromosome 7q, where the responsible gene, CLCN1, encodes the major skeletal muscle chloride channel. More than 70 mutations have been identified within CLCN1, and interestingly, some of these mutations are recognized to cause both dominant and recessive forms (Zhang et al., 2000). Examination of the functional effects of several myotonia-causing CLCN1 mutations in heterologous expression systems reveal effects on channel gating, usually resulting in a decreased chloride conductance (Zhang et al., 2000).
Potassium-Aggravated Myotonia
Pathophysiology
PAM links to chromosome 17q, where mutations in the SCN4A gene cause the disease. This sodium channel is the same one implicated in hyperKPP, PMC, and the sodium channel subtype of hypoKPP (see earlier discussion and Fig. 64.1). Functional expression studies reveal that the disease-causing mutations lead to a large persistent sodium current secondary to an increased rate of recovery from inactivation and an increased frequency of late channel openings (Wu et al., 2001). The cause of myotonia is this enhanced inward current, which leads to prolonged depolarization and subsequent membrane hyperexcitability.
Andersen-Tawil Syndrome
Clinical
Andersen-Tawil syndrome (ATS) is a rare autosomal dominant disorder characterized by the triad of periodic paralysis, cardiac arrhythmias, and dysmorphic features (including hypertelorism, micrognathia, low-set ears, high-arched or cleft palate, short stature, and clinodactyly) (Yoon et al., 2006a). The periodic paralysis, often triggered by rest after exercise, prolonged rest, and stress, is often the presenting symptom and can be hypo-, normo-, or hyperkalemic. The cardiac phenotype, often discovered later, includes prolonged QT intervals, but bidirectional ventricular tachycardia is common. Despite the known association of cardiac arrhythmias in rare periodic paralysis patients, ATS was only recognized as a separate entity in 1971. In families segregating an ATS allele, the phenotypic expressivity can vary greatly. Patients can manifest one, two, or three features of the triad, and the severity of any one feature can be extremely variable. Rare individuals are asymptomatic disease-gene carriers. Finally, ATS patients may exhibit neurocognitive deficits in executive function and abstract reasoning (Yoon et al., 2006b).
Pathophysiology
Mutations in the KCNJ2 gene on chromosome 17q account for approximately two-thirds of ATS probands. KCNJ2 encodes a widely expressed inwardly rectifying potassium channel (Plaster et al., 2001). Interestingly, among all identified probands, about 50% have an autosomal dominant disorder, and identification of sporadic cases with de novo mutations is common. The mechanisms of channel dysfunction are heterogeneous, including impaired phospholipid binding, pore function, or protein trafficking. Because VGKCs are tetrameric complexes, many (if not all) of the mutations are dominant negative.
Diagnosis
Previous studies that took into account the variable penetrance of ATS classified individuals as affected if two of three criteria were met: paroxysmal weakness, prolonged QT interval with or without ventricular dysrhythmias, or characteristic dysmorphic features (Yoon et al., 2006a). ATS should be included in the differential diagnosis of any individual with documented long-QT syndrome, even in the absence of periodic paralysis or dysmorphism. Some family members of patients with the full clinical triad show only prolonged QT intervals. Similarly, perform ECG on all patients with suspected periodic paralysis for careful measurement of the QT interval.




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


