Chapter 96 Channelopathies
Myotonic Disorders and Periodic Paralysis
The term channelopathies has been extended to include disorders affecting all organ systems, but this chapter focuses on channelopathies of skeletal muscle. The initial identification of channelopathies of skeletal muscle began with a number of diseases associated with mutations in the genes for specific channels, including the channels for chloride [Fialto et al., 2007; Trip et al., 2008; Davies and Hanna, 2003; Fahlke et al., 1997a, 1997b; George et al., 1993; Koch et al., 1992], sodium [Cannon, 1996; Struyk and Cannon 2007; Struyk et al., 2008; Fournier et al., 2004; Carle et al., 2009; Jurkat-Rott et al., 2009; Kubota et al., 2009; Matthews et al., 2008], calcium [Matthews et al., 2009; Urbano et al., 2008; Tomlinson et al., 2009], and potassium [Cannon, 2002; Sansone and Tawil; 2007; Sacconi et al., 2009] ions. Periodic symptoms are typical for most channelopathies and occur in disorders other than muscle diseases. For example, familial migraine and hereditary episodic ataxia result from mutations in the calcium channel. These two diseases have joined a growing list of disorders, including familial hypokalemic periodic paralysis, malignant hyperthermia, central core disease, and Lambert–Eaton myasthenic syndrome, that are also calcium channelopathies [Urbano et al., 2008]. This chapter limits its discussion to the disorders listed in Box 96-1 and refers the reader to the preceding references for discussion of other channelopathies.
Box 96-1 General Classification of the Channelopathies Seen in Children: Myotonias and Periodic Paralyses












The Myotonic Dystrophies
The myotonic dystrophies represent a group of dominantly inherited, multisystem (eye, heart, brain, endocrine, gastrointestinal tract, uterus, skin) diseases that share the core features of myotonia, weakness, and early-onset cataracts (younger than 50 years). Table 96-1 summarizes myotonic dystrophy types 1 and 2. Clinicians considered myotonic dystrophy to be a single disease up until the mid-1990s. Steinert and colleagues described the disorder clearly in the early 1900s and that description is still valid [Harper, 2001]. The gene defect responsible for myotonic dystrophy of Steinert was discovered in 1992 and found to be caused by expansion of a CTG repeat in the 3′ untranslated region of DMPK, a gene encoding a protein kinase [Brook et al., 1992; Fu et al., 1992; Mahadevan et al., 1992]. After the discovery of this gene defect, DNA testing revealed a group of patients with dominantly inherited myotonia, proximal greater than distal weakness, and cataracts, who previously were diagnosed as having myotonic dystrophy but who, after testing, lacked a CTG repeat expansion on the DMPK gene. Subsequent clinical studies of kindreds with patients having these characteristics led to the use of new diagnostic labels for these patients: myotonic dystrophy type 2 [Thornton et al., 1994] and proximal myotonic myopathy (PROMM) [Ricker et al., 1994a]. Later studies demonstrated that many of the families identified as having myotonic dystrophy type 2 or PROMM had the same disease, a disorder that results from an unstable four-nucleotide repeat expansion (CCTG) on chromosome 3 [Liquori et al., 2001]. This disease is similar to, but distinct from, classic myotonic dystrophy of Steinert.
Table 96-1 Myotonias Due to Unstable Nucleotide Repeat Expansion
| Clinical Features | Myotonic Dystrophy of Steinert (Myotonic Dystrophy Type 1, DM1) | Myotonic Dystrophy Type 2 (Proximal Myotonic Dystrophy, DM2) |
|---|---|---|
| Inheritance | Dominant | Dominant |
| Gene defect | Chromosome 19; CTG expansion affecting a protein kinase; repeat size ranges from 50 to >2000; normal 5–37 repeats | Chromosome 3; CCTG expansion affecting the zinc finger 9 protein gene; repeat sizes vary from 78 to >11,000; normal <78 repeats |
| Age of onset | Broad range of ages (infancy to adulthood), with infant onset in most severe cases | Broad range of ages (late childhood to late adulthood) |
| Myopathy | Face, eyes, forearm, hands, and legs, with generalized weakness and hypotonia in affected infants | Mild; thighs, hips, neck flexors, occasional calf muscle, abdominal muscle hypertrophy |
| Myotonia | Primarily affects hand and forearm muscles and tongue; occasionally affects respiratory muscles and smooth muscle, such as intestine or uterus; myotonia improves with heat and exercise; myotonia results from a decrease in chloride channel protein | Mainly in hands and thighs; varies; frequently hard to detect; pain occurs sometimes with and without myotonia; myotonia improves with repeated contractions; myotonia results from a decrease in chloride protein |
| Provocative stimuli | Myotonia worsened by rest and cold; myotonia is relatively constant in severity and muscles affected | Myotonia worsened by rest but varies in severity, occasionally being absent on clinical examination; hand grip and thigh stiffness are usual sites |
| Therapy for symptoms | Bracing; cataract removal; monitoring dysrhythmias and respiratory insufficiency; pacemaker; antimyotonia therapy (mexiletine); avoid depolarizing muscle relaxants, opiates, and barbiturates with surgery | Cataract removal; occasional need for pacemaker; antimyotonia therapy often not necessary (mexiletine); monitor carefully during and after surgery for muscle rigidity and rhabdomyolysis |
The existence of different types of myotonic dystrophy has created a need to develop a diagnostic classification. To address this need, the International Myotonic Dystrophy Consortium developed a new nomenclature and guidelines for DNA testing [IDMC, 2000]. Myotonic dystrophy of Steinert, the classic form of myotonic dystrophy that results from an unstable trinucleotide repeat expansion on chromosome 19, is now termed myotonic dystrophy type 1 (DM1). Patients with the clinical picture of myotonic dystrophy type 2/proximal myotonic myopathy (DM2), who have positive DNA testing for the unstable four-nucleotide repeat expansion on chromosome 3, are now classified as having DM2 [Day et al., 2003; Thornton et al., 1994].
Myotonic Dystrophy Type 1
Clinical Features and Subclassification
DM1 is a multisystem, autosomal-dominantly inherited, highly variable muscle disease with an incidence of 1 in 8000 individuals (see Table 96-1) [Harper, 2001; Moxley and Meola, 2008]. Childhood-onset DM1 may present as a severe early congenital form, or as a milder form with symptoms beginning in the early school years. The congenital form presents at birth with respiratory failure, poor feeding, generalized hypotonia, clubfoot deformity, and an increased risk of intracerebral hemorrhage and eventration of the diaphragm [Harper, 2001]. During gestation, there are often reduced fetal movements and a history of polyhydramnios [Harper, 2001; Zaki et al., 2007]. There is an increased frequency of placenta previa and miscarriage [Harper, 2001; Rudnik-Schoneborn and Zerres, 2004; Rudnik-Schoneborn et al., 2006], and, not uncommonly, a failed labor may require urgent cesarean section. During the perinatal period, infants with congenital myotonic dystrophy may require continuous ventilator support, and those remaining ventilator-dependent beyond 4 weeks of age have a poor prognosis for survival, although recent advances in neonatal intensive care have allowed many affected infants to survive, with subsequent slow improvement in function throughout childhood and teens [Campbell et al., 2004; Harper, 2001]. During the first 2 years of life, children with congenital myotonic dystrophy are at increased risk for aspiration pneumonitis and difficulties with feeding. Mental retardation and learning disabilities are common in congenital myotonic dystrophy. Determining the degree of cognitive deficit requires careful evaluation because these patients have marked facial weakness and are unable to speak and communicate well until mid- to late childhood. There is also an increased incidence of hearing loss [Harper, 2001]. These problems often complicate neuropsychologic testing. Figure 96-1A shows a mother with two daughters, both of whom have congenital myotonic dystrophy. Both children have delayed ability to speak and cognitive impairment. Their mother has mild ptosis, minimal grip myotonia, and minimal distal weakness. She is fully employed and has no cognitive impairment. This mother has the classic adult-onset form of DM1 [Harper, 2001].

The later, childhood-onset form usually is evident from nonmuscular manifestations of the disease, such as intellectual deficiency, difficulty with speech or hearing, clumsiness, or, rarely, with a cardiac dysrhythmia or postoperative apnea [Harper, 2001; Sinclair and Reed, 2009]. DM1 patients may have an increased sensitivity to sedative medications, especially barbiturates and opiates. and occasionally experience increased muscle stiffness/myotonia during general anesthesia [Harper and van Engelen, 2004; Sinclair and Reed, 2009]. Cardiac arrhythmias are more common complications of the adult-onset forms of DM1 [Breton and Mathieu, 2009; Groh et al., 2008; Harper, 2001]. However, other nonmuscular manifestations, such as gastrointestinal hypomotility with chronic constipation, and intermittent urinary tract symptoms, such as urgency or frequent urination, are common in childhood myotonic dystrophy [Harper, 2001; Harper and van Engelen, 2004].
Genetics
DM1 results from an unstable trinucleotide expansion in the 3′ untranslated region of a gene on chromosome 19 that codes for a serine/threonine protein kinase, DMPK [Brook et al., 1992; Fu et al., 1992; Mahadevan et al., 1992]. The exact function of this kinase remains unclear. The cause of the unstable CTG repeat expansion also is unknown. Recent research suggests that premutations arise from the upper normal range of CTG repeat region sizes and become pathologically enlarged within a few generations [Abbruzzese et al., 2002; Martorell et al., 2001]. Past investigations demonstrate that mitomycin C can enhance enlargement of pathologically expanded repeats and promote enlargement of normal repeats [Pineiro et al., 2003]. Inhibitors of replication also modulate instability of the CTG repeat in the DM1 gene in cultured cells [Yang et al., 2003]. These observations raise the possibility that environmental/external factors may influence the development of pathologic enlargement of CTG repeats at the DM1 locus. Recent investigations suggest that DNA “repair” proteins may also have a mutagenic role in CTG repeat instability [Slean et al., 2008; Pearson et al., 2005]. The normal size of the [CTG]n repeats ranges from 5 to 37 repeats. Children with the congenital form of DM1 have expansions typically greater than 1500 repeats [Harper, 2001]. Moderately affected adults with DM1 have [CTG]n sizes ranging from 300 to 1000 repeats, and mildly affected individuals have expansions from 50 to 200 repeats. There are no examples of point mutations in the DMPK gene locus that have caused the clinical picture of DM1. The same is true for DM2.
The unstable expansion of the DM1 gene provides an explanation for the well-described phenomenon of anticipation: that is, the earlier onset of more severe manifestations of disease in successive generations. Figure 96-1 emphasizes the phenomenon of anticipation in the mother with mild to moderate DM1 has had two children with severe congenital DM1.
Pathophysiology
The likely molecular pathomechanism that underlies both DM1 and DM2 relates to the nuclear accumulation of RNA containing the abnormally expanded repeats (CUG in DM1 and CCUG in DM2). These accumulations of RNA are unable to exit from the nucleus and undergo normal removal [Osborne et al., 2009; Wheeler, 2008]. The abnormal accumulation of the expanded repeat-containing RNA alters the normal functions of specific nuclear regulatory proteins, such as muscleblind [Mankodi and Thornton, 2002; Mankodi et al., 2003] and CUGBP1 [Timchenko et al., 2002; Savkur et al., 2001; Charlet et al., 2002]. The net effect is a novel toxic RNA disease mechanism that causes abnormal splicing of pre-mRNA in both DM1 and DM2 [Osborne et al., 2009; Wheeler, 2008]. The toxic RNA hypothesis builds heavily upon studies of animal models, as well as on findings in human tissues. The normal DMPK gene contains a short CTG repeat in the 3′ untranslated region of exon 15, and expression of this gene produces an mRNA containing a short CUG repeat [Wheeler, 2008]. The normal zinc finger protein 9 gene (ZNF9 gene), which contains, in intron 1, a large CCTG expansion that is responsible for DM2 [Liquori et al., 2001], and normally contains a short CCTG repeat in intron 1, gives rise to a pre-mRNA containing a CCUG repeat [Wheeler, 2008]. Due to removal of the introns during the editing of pre-mRNA, the CCUG repeat tract is absent from the ZNF9 mRNA [Margolis et al., 2006]. In the presence of nonexpanded CUG and CCUG repeats, the activities of muscleblind nuclear regulatory protein 1 (MBNL1) and the CUGBP1 protein are normal [Wheeler, 2008]. This allows normal regulation of pre-mRNA splicing by these nuclear regulatory proteins and normal production of the proper proteins in the cell [Wheeler, 2008].
The misregulation that occurs in DM1 and DM2 causes a “spliceopathy” that affects at least two-dozen transcripts [Osborne et al., 2009; Wheeler, 2008]. Myotonia caused by a marked reduction in the synthesis of the adult form of the skeletal muscle chloride channel [Charlet et al., 2002; Mankodi et al., 2002], and insulin resistance caused by deficient synthesis of the adult form of the insulin receptor [Savkur et al., 2001, 2004] occur in DM1 and DM2 as a result of this splicing defect, and it is likely that additional disease manifestations result from this alteration. In DM1, not DM2, excess phosphorylation of CUGBP1 occurs and results in higher total levels of this protein, gain of function, and in turn perhaps leads to the more pronounced muscle wasting often seen in DM1 [Wheeler, 2008].
The cause of the muscle wasting and weakness in DM1 remains a mystery. In the transgenic mouse model developed by Mankodi and colleagues, the mice do not develop muscle wasting or significant weakness [Mankodi et al., 2000, 2002]. In DM1 patients, there are hormonal abnormalities, including testosterone deficiency, insulin resistance, hyperinsulinemia, and altered release of growth hormone, which may contribute to the muscle wasting. To date, however, therapeutic trials restoring or exceeding physiologic levels of these hormones have failed to reverse weakness and wasting [Harper, 2001; Moxley et al., 2004].
Recently, two new mouse models have been developed and provide encouraging new discoveries related to skeletal muscle wasting and cardiac disease in DM1. One study describes an inducible mouse model of severe skeletal muscle wasting that expresses large tracts of CTG repeats in DMPK exon 15 [Orengo et al., 2008]. The results indicate that increased CUGBP1 protein levels are associated with increased DMPK-CUG RNA expression, and suggest a role for CUGBP1-specific splicing or cytoplasmic functional alterations as causes of muscle wasting [Orengo et al., 2008]. The investigators found that pentamidine partially rescues the defect in splicing of two pre-mRNAs (the skeletal muscle chloride channel and Serca1 pre-mRNA) in this mouse model [Orengo et al., 2008], raising the possibility of future therapeutic trials with pentamidine.
Another recent study demonstrates that, in addition to the sequestration of MBNL1 by the toxic RNA CUG repeat tract in DM1, these expanded CUG tracts activate the protein kinase C (PKC) signaling pathway, which leads to hyperphosphorylation and stabilization of CUGBP1 in DM1 heart and skeletal muscle tissues [Wang et al., 2009]. This elevation of hyperphosphorylated CUGBP1 may have an important pathophysiological role in causing cardiac muscle dysfunction in DM1. To explore this possibility, the investigators used an inducible DM1 mouse model, in which a transgene containing the last exon of DMPK with 960 CTG repeats is induced to express CUG repeat-containing RNA [Wang et al., 2009]. These mice exhibited higher mortality, conduction abnormalities, and systolic and diastolic dysfunction, as well as molecular changes typical of patients with DM1. One group of these mice received PKC inhibitors and exhibited increased survival that correlated with decreased phosphorylation and decreased steady-state levels of CUGBP1. Treatment with PKC inhibitors also reduced misregulation of splicing that was normally regulated specifically by CUGBP1, but not those splicing events regulated by MBNL1 [Wang et al., 2009]. The investigators suggest that pharmacological blockade of PKC activity mitigates the DM1 cardiac phenotype and they propose that the PKC pathway plays an important role in the pathogenesis of DM1. More investigation is necessary to confirm and extend these findings.
Clinical Laboratory Tests
Identification of the abnormal expansion of [CTG]n repeats in the DMPK gene on chromosome 19 establishes the diagnosis of DM1. Electromyography to detect myotonia is helpful in older children, but myotonia may be absent in infancy or in early childhood. Slit-lamp examination to identify the typical iridescent, spokelike posterior capsular cataracts is helpful. Muscle biopsy is not necessary to diagnose DM1 but may be useful to exclude other disorders. Careful examination of the mothers of patients with suspected congenital DM1 may reveal subtle clinical findings. Muscle pathology often demonstrates prominent central nuclei, scattered angular fibers, pyknotic nuclear clumps, and type 1 fiber atrophy. Creatine kinase levels may be normal or elevated and are not of primary value in the diagnosis [Heatwole et al., 2006]. With the discovery of the gene defect in DM1, it is possible to perform both prenatal and perinatal genetic counseling. Analysis of the DNA isolated from amniocytes or chorionic villus samples can predict the delivery of affected infants and the severity of illness [Harper, 2001; Harper and van Engelen, 2004].
Treatment
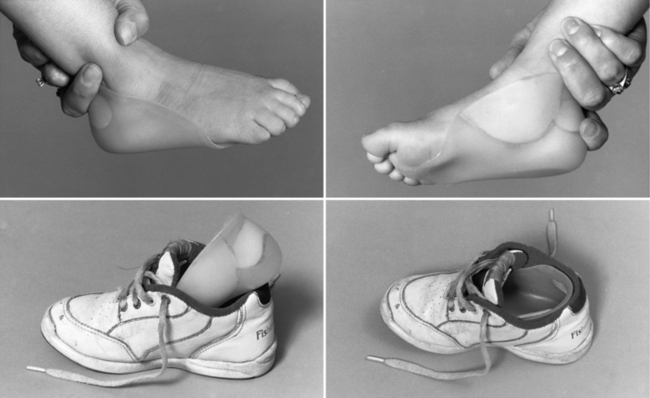
Treating patients with congenital myotonic dystrophy involves ventilatory support, use of a feeding tube, bracing for clubfoot deformities when present, and occasional corrective surgery for foot deformities (Figure 96-2) [Harper, 2001; Harper and van Engelen, 2004; Canavese and Sussman, 2009]. Figure 96-2 shows the molded plastic orthosis that helps treat foot deformity later in childhood. Speech problems and abdominal pain may improve with antimyotonia treatment. Mexiletine is sometimes helpful in alleviating some of the symptoms, especially in later childhood or adolescence when patients may have recurrent dislocation of the mandible, and pain and muscle spasm in the masseter muscles. Electrocardiographic monitoring is necessary on a regular basis, particularly during treatment with mexiletine, to search for covert dysrhythmias [Christensen et al., 2008; Otten et al., 2009; Gorog et al., 2005]. Treatment should be coordinated with the child’s school to ensure that cognitive deficiencies and hearing deficits are monitored. The increased risk for cardiac dysrhythmia and apnea after administration of general anesthesia for surgery requires overnight hospitalization with monitoring [White and Bass, 2003; Sinclair and Reed, 2009]. Apnea can develop several hours after a patient has been extubated [Harper and van Engelen, 2004]. No specific limitation on physical activity is necessary during childhood, provided the child has gained sufficient strength and coordination to carry out activities requested in the school environment.
Myotonic Dystrophy Type 2 (Formerly Proximal Myotonic Myopathy)
Clinical Features
DM2/PROMM typically appears in adult life and has variable manifestations, such as early-onset cataracts (younger than 50 years), varying grip myotonia, thigh muscle stiffness, and muscle pain, as well as weakness (hip flexors, hip extensors, abdominal muscles, or long flexors of the fingers) [Day et al., 2003; Moxley et al., 2002; Ricker et al., 1994a, 1995; Schoser et al., 2004b; Thornton et al., 1994]. Symptoms often appear between 20 and 70 years of age, and patients, as well as their care providers, ascribe them to overuse of muscles, “pinched nerves,” “sciatica,” arthritis, fibromyalgia, or statin use [George et al., 2004]. Younger patients may complain of stiffness or weakness when running up steps. In general, DM2 muscle pain has no consistent relationship to exercise or to the severity of myotonia found on clinical examination. The pain, which tends to come and go without obvious cause, usually fluctuates in intensity and distribution over the limbs. It can last for days to weeks. This pain seems qualitatively different from the muscle and musculoskeletal pain that occurs in patients with DM1.
Early in the presentation of DM2 there is only mild weakness of hip extension, thigh flexion, and finger flexion. Myotonia of grip and thigh muscle stiffness vary from minimal to moderate severity over days to weeks. Direct percussion of forearm extensor and thenar muscles is the most sensitive clinical test for myotonia in DM2 but may be absent. Myotonia of grip is sometimes prominent and often has a jerky quality that seems to differ from that in DM1 and the nondystrophic myotonias. Myotonia is often less apparent in DM2, compared to patients with DM1. In cases of late-onset DM2, myotonia may only appear on electromyographic testing after examination of several muscles [Day et al., 2003; Ricker et al., 1995]. Facial weakness is mild in DM2, as is muscle wasting in the face and limbs. Weakness of neck flexors is frequent. Trouble arising from a squat is common, especially as the disease progresses. Calf-muscle hypertrophy occasionally is prominent [Ricker et al., 1994a, 1995; Thornton et al., 1994]. Other manifestations, such as excessive sweating, hypogonadism, glucose intolerance, dysphagia, cardiac conduction disturbances, and cognitive/ neuropsychologic alterations, may also occur and worsen over time [Day et al., 2003; Meola et al., 1999, 2003; Schoser et al., 2004c]. A higher rate of premature labor and preterm deliveries also has been reported [Rudnik-Schoneborn et al., 2006].
At present there is no clear evidence of a congenital form of DM2 [Day et al., 2003]. However, there is evidence of anticipation, with earlier onset of more prominent symptoms, in studies of parent-child pairs [Schneider et al., 2000]. When symptoms occur in childhood, they typically are transient and relate to grip myotonia in the hands and thighs. Proximal weakness, muscle pain, cataracts, and occasional cardiac dysrhythmias usually do not develop until mid- or late adult life [Ricker et al., 1995; Thornton et al., 1994].
Genetics
DM2 results from an unstable four-nucleotide repeat expansion, CCTG, in intron 1 of the zinc finger protein 9 gene on chromosome 3q21 [Bachinski et al., 2003; Liquori et al., 2001, 2003]. The cause of the unstable expansion is unknown. The size of the CCTG repeat appears to increase over time in the same individual, and, like DM1, it is a dynamic gene defect [Day et al., 2003]. The size of the CCTG repeat in normal leukocyte DNA is less than 75 repeats [Day et al., 2003; Liquori et al., 2001]. The size in DM2 ranges from 78 to over 11,000 repeats [Day et al., 2003; Liquori et al., 2001]. The gene mutation responsible for DM2 appears to have arisen from a northern European founder [Bachinski et al., 2003; Liquori et al., 2003].
Pathophysiology
The molecular mechanisms leading to the manifestations of DM2 are believed to be similar to that in DM1, and relate to a toxic effect of the abnormally expanded RNA that accumulates in the muscle nuclei (see previous section on Pathophysiology of DM1). There is no apparent relationship between the putative functions of the zinc finger protein 9 gene at 3q21 and the DM1 gene at 19q13.3 or their gene products [Liquori et al., 2001]. Flanking genes are not similar for the DM2 and DM1 loci. A toxic effect of the ribonucleic acid made from the unstable, abnormally expanded, nucleotide repeats in the DM2 and DM1 genes is likely to serve as the common mechanism for their disease manifestations.
The cause of the weakness in DM2 is unclear. Patients with DM2, in contrast to patients with classic DM1, usually have only mild muscle wasting. However, there is an uncommon, adult-onset variant of DM2, termed proximal myotonic dystrophy [Udd et al., 1997], which causes severe wasting of proximal arm and thigh muscles as the illness progresses. Patients with the proximal myotonic dystrophy variant of DM2 have the identical CCTG repeat expansion at 3q21 that occurs in patients with DM2 [Bachinski et al., 2003]. The reason for the severe muscle wasting in this variant, compared with DM2 patients, is unknown. Whether individuals with proximal myotonic dystrophy [Udd et al., 1997] share an underlying mechanism responsible for muscle wasting that is similar to the mechanism in DM1 remains to be established. Two recent reports using cell models describe a reduction in the rate of protein translation in DM2 muscle cells, and propose that the accumulation of RNA containing long tracts of CCUG dysregulates translation and degradation of proteins in patients [Salisbury et al., 2009; Huichalaf et al., 2009]. More research is needed to confirm and extend these initial observations.
Clinical Laboratory Tests
Leukocyte DNA testing is available for DM2. DNA testing methodology used in the recent past to screen for DM2 may have failed to detect as many as 20 percent of affected individuals [Day et al., 2003]. Current methods use the more sensitive DNA testing methodologies for DM2 [Bachinski et al., 2003; Day et al., 2003].
Patients with elevated creatine kinase values may have greater postoperative complications [Ricker et al., 1995]. A recent comparison of muscle biopsy findings in DM2 with those in classic DM1 indicates that there is less severe atrophy of fibers in DM2, and that in DM2 there are subgroups of very small type 2 fibers and nuclear clumps [Vihola et al., 2003]. Both DM2 and DM1 display abundant central nuclei on their biopsies [Day et al., 2003; Harper, 2001; Vihola et al., 2003], but it appears that DM2 is predisposed to type 2 fiber atrophy and DM1 to type 1 fiber atrophy [Cardani et al., 2009; Pisani et al., 2008; Bassez et al., 2008; Schoser et al., 2004a; Vihola et al., 2003].
Treatment
In general, the management of DM2 is similar to that of DM1, but there is less need for supportive care, such as bracing, scooters, or wheelchairs. Cataracts require monitoring, and serial monitoring with an electrocardiogram (EKG) is necessary to check for covert dysrhythmia. Disturbances in cardiac rhythm are less frequent in DM2, but abnormalities do occur [Wahbi et al., 2009; Schoser et al., 2004b; Day et al., 2003; Moxley et al., 2002]. Hypogonadism and insulin resistance need monitoring, as in DM1. Myotonia tends to be less marked and less troublesome in DM2 but, in specific circumstances, antimyotonia therapy is helpful, especially if muscle stiffness is frequent and persistent or if pain is prominent. Cognitive difficulties also occur in DM2, as in DM1, but become manifest in adult life and appear to be associated with decreased cerebral blood flow to frontal and anterior temporal lobes [Meola and Sansone, 2007; Meola et al., 1999] and decreased brain volume [Akiguchi et al., 1999; Chang et al., 1998]. The changes are less severe than in DM1. Their etiology is unknown but may relate to the toxic effect of intranuclear accumulations of abnormally expanded RNA. Management of these brain symptoms is similar to that for DM1.
A frequent and difficult problem in DM2 is the peculiar muscle pain described earlier [Auvinen et al., 2008; George et al., 2004]. The exact mechanism underlying the pain is unknown, and there is no well-established, effective treatment. Carbamazepine or mexiletine, along with nonsteroidal anti-inflammatory medications or acetaminophen ameliorates this pain in some patients.
Experimental Therapeutics in Myotonic Dystrophy
Evaluating the differences and similarities between DM1 and DM2, both clinically and in their molecular mechanisms, presents opportunities for the development of potential new treatments and provides hope to researchers and patients that treatment trials may not be far away. Support for this optimistic statement comes from recent studies in mouse models of DM1 that include a demonstration of reversal of the spliceopathy. Treatment of transgenic mice that over-express CUG transcripts containing 250 repeats with an antisense 25-nucleotide morpholino oligonucleotide composed of CAG repeats led to the dispersal of the nuclear foci that contained accumulations of toxic RNA and freed up the nuclear regulatory protein, MBNL1 [Wheeler et al., 2009]. This treatment also reversed the spliceopathy and led to restoration of normal synthesis of the proper amount of the adult form of the skeletal muscle chloride channel. Myotonia also disappeared in the treated mice [Wheeler et al., 2009]. In another study, researchers used a myoblast-myotube cell model system of DM1 cells containing 300 CTG repeats and found that treatment with a 2′-O-methyl-phosphorothioate-modified (CAG)7 antisense oligonucleotide silenced mutant DMPK RNA expression and reduced the number of ribonuclear aggregates [Mulders et al., 2009]. Use of this same antisense oligonucleotide in vivo in a DM1 mouse model caused a significant reduction in the level of toxic (CUG) RNA and corrected the defect in splicing of pre-mRNA [Mulders et al., 2009]. In the future, it may be possible to develop a double-pronged therapeutic approach that combines antisense oligonucleotide treatment to free up sequestered MBNL1 nuclear regulatory protein from the toxic CUG repeat tract, and another antisense oligonucleotide that facilitates degradation of toxic RNA. Antisense oligonucleotides are already in clinical trials to treat other diseases, including Duchenne muscular dystrophy and familial hypercholesterolemia, and DM1 may soon be added to this list.
Autosomal-Dominant and Autosomal-Recessive Myotonia Congenita
Thomsen’s disease (autosomal-dominant myotonia congenita) and Becker’s disease (autosomal-recessive myotonia congenita) represent two forms of chloride channelopathies (Table 96-2). The chloride channelopathies that produce myotonia as their primary symptom resemble the sodium channel disorders without periodic paralysis, and may resemble mild forms of DM1 and DM2. Other causes of muscle stiffness or poor coordination, including central nervous system diseases affecting the frontal lobes, brainstem, and cerebellum, require initial consideration but are confused rarely with these channelopathies.
Table 96-2 Chloride Channel Myotonias
| Clinical Features | Autosomal-Dominant Myotonia Congenita of Thomsen | Autosomal-Recessive Generalized Myotonia of Becker |
|---|---|---|
| Inheritance | Dominant | Recessive |
| Gene defect | Chromosome 7; mutation in skeletal muscle chloride channel | Chromosome 7; mutation in skeletal muscle chloride channel |
| Age of onset | Infancy to early childhood | Late childhood, occasionally starts earlier or begins in teens |
| Myopathy | Muscle hypertrophy frequent; no myopathy, although variants uncommonly develop weakness | Occasional muscle wasting and weakness can occur late; hypertrophy of muscles frequently occurs in legs |
| Myotonia | Generalized stiffness, especially after rest; improves with exercise; prominent myotonia of eye closure, but not paradoxical myotonia | Generalized stiffness, especially after rest; transient weakness is prominent after complete relaxation for several minutes; myotonia occurs in eyes; no paradoxical myotonia |
| Provocative stimuli | Prolonged rest or maintenance of the posture | Prolonged rest or maintenance of the same posture |
| Therapy for symptoms | Exercise; antimyotonia therapy (e.g., mexiletine); Achilles’ tendon stretching helps prevent need for heel cord-lengthening surgery | Exercise; especially avoiding prolonged rest; antimyotonia therapy (e.g., mexiletine); transient weakness does not improve after mexiletine |
Clinical Features
Generalized myotonia is the major clinical symptom in dominant and recessive forms of chloride channel myotonia congenita. Recent studies indicate that pain occurs in almost 30 percent of patients with chloride channel myotonia [Trip et al., 2009a], and as many as 75 percent have mild generalized muscle weakness that is slightly greater proximally [Trip et al., 2009a; Shalata et al., 2009; Bernard et al., 2008]. The pain has a close relation to fatigue and has a negative impact on the health status of those affected [Trip et al., 2009a]. Symptoms develop in the first or second decade of life. Myotonic stiffness occurs with sudden physical exertion after a period of rest. Repeated muscle contractions ameliorate the stiffness. This response is the “warm-up phenomenon,” which helps distinguish chloride channel myotonia congenita from certain forms of sodium channel myotonia that display increasing muscle stiffness with repeated contractions, a paramyotonic response. DM1 and DM2, like chloride channel myotonia, demonstrate the typical warm-up response.
In contrast to Thomsen’s disease, patients with autosomal-recessive myotonia congenita (Becker’s disease) have transient proximal muscle weakness. The transient weakness appears for a few seconds during the initial attempt at a specific movement after a period of inactivity [Zwarts and VanWeerden, 1989]. Muscle strength improves to normal after several strong contractions [Baumann et al., 1996; Zwarts and VanWeerden, 1989]. One study suggests that this transient weakness represents a transient depolarization block associated with prolonged recovery of muscle membrane excitability [McKay et al., 2006].
Genetics
Point mutations in the gene for the skeletal muscle chloride channel cause both autosomal-dominant [Trip et al., 2008, 2009b; Fialho et al., 2007; Fahlke et al., 1997a; Koch et al., 1992] and autosomal-recessive [Trip et al., 2008, 2009b; Fialho et al., 2007; Koch et al., 1992] forms of myotonia congenita. Autosomal-dominant myotonia congenita (Thomsen’s disease) may vary considerably in age of onset and severity of myotonia [Duno et al., 2004; Koty et al., 1996]. Two recent reports describe marked myotonia, generalized moderate weakness, and generalized muscle hypertrophy in homozygous individuals in kindreds with dominant chloride channel myotonia [Bernard et al., 2008; Shalata et al., 2009]. These results demonstrate a dose effect with disease severity. It is now possible to screen for known point mutations in the chloride channel gene on chromosome 7 [Trip et al., 2008, 2009b; Fialho et al., 2007], and extensive genetic testing for more than 80 mutations is available in two laboratories, one in the United Kingdom and one in the Netherlands. The group in the Netherlands recommends a tandem analysis for gene mutations. They first perform DNA analysis of all 23 exons in the skeletal muscle chloride channel gene, and then proceed with DNA analysis of those individuals who test negative for chloride channel mutations, with probing of the exons in the skeletal muscle sodium channel gene known to cause sodium channel myotonia [Trip et al., 2008]. This approach has proven very informative in establishing a diagnosis.
Pathophysiology
Electrophysiologic studies in myotonic goats [Lipicky and Bryant, 1993] and human muscle tissue from patients with autosomal-dominant and autosomal-recessive [Koch et al., 1992; Rudel et al., 1994] myotonia congenita demonstrate a decreased chloride conductance. The decreased chloride conductance across the transverse tubular system renders the muscle membrane hyperexcitable, which leads to after-depolarization and repetitive firing, creating clinical myotonia. Both autosomal-dominant myotonia congenita (Thomsen’s disease) and generalized autosomal-recessive myotonia congenita (Becker’s disease) involve mutations at the same locus [George et al., 1993; Koch et al., 1992]. Current models for the chloride channel indicate that two subunits combine to form a double-barreled entry gate for chloride ions and a single outflow channel [Duffield et al., 2003; Fahlke et al., 1997a, 1997b; Saviane et al., 1999]. Dominant mutations affect the common outflow channel (slow-flow channel), and recessive mutations require involvement of the two faster-flow entry gates [Duffield et al., 2003]. The model proposes that involvement of only one entry gate does not cause clinical disease. Recessive mutations cause a pathologic reduction in chloride current because both fast gates undergo blockade, and dominant mutations cause pathologically reduced current because they affect the slower common exit channel for chloride ions. Recessive mutations are much more frequent than dominant mutations. Over 80 mutations are known to cause chloride channel myotonia [Colding-Jorgensen, 2005]. More than 50 percent of chloride channel mutations occur in exons 3, 8, and 11 [Fialho et al., 2007; Trip et al., 2008]. The variations in the clinical severity of certain very rare, dominantly inherited forms of chloride channel myotonia, such as “myotonia levior” [Ryan et al., 2002] and “fluctuating myotonia” [Wagner et al., 1998], are interesting, and point to limitations in our current version of the model as an explanation for the clinical spectrum of findings. A more recent report further emphasizes the limitation of the model. This report describes two unrelated families with the Becker recessive form and two unrelated families with the Thomsen dominant form of myotonia congenita, all of whom have a common R894X mutation [Duno et al., 2004]. The dominant family with the most severe phenotype expressed twice the expected amount of the R894X mRNA allele. These findings suggest that allelic variation may be an important modifier of disease progression in myotonia congenita. One report of a family with Thomsen myotonia congenita describes a kindred with a T310M mutation and notes that family members with one mutated allele have mild manifestations, while the one family member who was homozygous for this mutation had severe stiffness, generalized muscle hypertrophy, and moderate weakness [Bernard et al., 2008]. Another more recent report also describes a gene dose effect, with worse disease severity (i.e., more severe myotonia and generalized weakness) in homozygous individuals who have a missense mutation of glycine to serine (G190S) in a well-conserved motif in helix D of the skeletal muscle chloride channel gene [Shalata et al., 2009].
The model for the chloride channel also does not provide an explanation for the transient weakness that is a prominent clinical feature in generalized autosomal-recessive (Becker’s myotonia) myotonia congenita. Its cause remains to be discovered. The model fails, as well, to provide an explanation for transient worsening that occurs occasionally during pregnancy. One potential clue comes from recent in vitro studies of testosterone and progesterone, which show rapid and reversible inhibition of both the normal and the mutated forms of skeletal muscle chloride channels expressed in Xenopus oocytes, caused by a prominent rightward shift in the voltage dependence of their open probability [Fialho et al., 2008]. Further study using more physiologic concentrations of these sex hormones in a more appropriate cell culture model, such as myoblasts or myotubes, is needed to establish the physiological significance of this effect.
Clinical Laboratory Tests
Distinguishing patients with chloride channel myotonia, either autosomal-dominant or autosomal-recessive forms, from those with sodium channel myotonic disorders is more difficult. Both cause myotonic stiffness and muscle hypertrophy, especially in the legs. One distinguishing feature of sodium channel myotonia is the paradoxical myotonia of the eyelids that develops with repeated forceful eye closure. Figure 96-3 shows the response to looking up and down and to eye closure in a patient with DM1. The patient in Figure 96-4 has sodium channel myotonia, exhibiting lid lag and persisting myotonia after forceful closure of the eyes. If this patient repeats the forceful closure of his eyes, his myotonia worsens. Persistent myotonia of the eyelids also occurred in this patient’s son. In contrast, patients with autosomal-dominant and autosomal-recessive forms of chloride channel myotonia exhibit warm-up after repeated contractions. One recent study comparing 32 patients with chloride channel myotonia to 30 patients with sodium channel myotonia observed the following differences between these two groups of patients [Trip et al., 2009b]. Compared to patients with sodium channel myotonia, patients with chloride channel myotonia have a higher frequency of: muscle weakness, 75 versus 37 percent; warm-up following repeated 3-second isotonic maximum grips, 100 versus 47 percent; and slowing in the time to arise from sitting to standing, 91 versus 67 percent. In contrast, compared to patients with chloride channel myotonia, patients with sodium channel myotonia had higher frequencies of: paradoxical myotonia, 50 versus 0 percent; and painful myotonia, 57 versus 28 percent [Trip et al., 2009b]. Patients with sodium channel myotonia also had earlier onset of symptoms: 4.4 years versus 10 years of age. Patients with chloride channel myotonia do not have a worsening of myotonic stiffness or paralysis after prolonged exposure of muscle to cold. To search for cold-induced paralysis, it is necessary to soak the hand or forearm muscles in cold water (15°C) for 15–20 minutes. If there is weakness with exercise after this cold exposure, this observation strongly favors sodium channel myotonia rather than chloride channel disease [Heatwole and Moxley, 2007; Carle et al., 2009; Webb and Cannon, 2008]. The subsequent section on sodium channel myotonia briefly describes the laboratory evaluation of the sodium channelopathy disorder, hyperkalemic periodic paralysis. Recent reports have provided protocols for complete screening of both the skeletal muscle chloride and sodium channel genes [Trip et al., 2008; Fialho et al., 2007]. The approach uses tandem analysis of the chloride channel gene, followed by analysis of the sodium channel gene, in individuals who already have had negative DNA analysis for DM1 and DM2, and have a negative history for periodic paralysis.

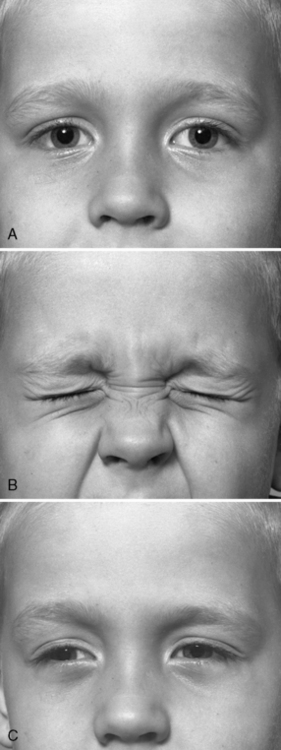
Fig. 96-4 A patient with acetazolamide-responsive sodium channel myotonia.
The patient appears in Figure 96-1 with his father, who is also shown in Figure 96-3. Sequential photographs of the patient before and after forced closure of the eyes. Myotonia appears and worsens after repeated eye closure (e.g., paramyotonia).
Treatment
Antimyotonia treatment with mexiletine is often helpful [Heatwole and Moxley, 2007]. Before treatment with mexiletine is initiated, a baseline EKG is necessary to identify patients with unsuspected cardiac conduction abnormalities. Side effects of mexiletine, which are usually mild and dose-related, include dysgeusia, lightheadedness, and diarrhea. Tocainide, another lidocaine derivative similar to mexiletine, was another useful alternative [Kwiecinski et al., 1992], but it is no longer available. Isolated reports suggest that carbamazepine may be a readily available alternative antimyotonia medication for myotonia congenita [Berardinelli et al., 2000; Sheela, 2000]. Children with moderately severe myotonia develop heel-cord shortening and contractures at their elbows. If these contractures do not respond to stretching and other physical therapy exercises, it is appropriate to initiate antimyotonia treatment, even when patients do not complain of stiffness.
Acetazolamide-Responsive Sodium Channel Myotonia and Myotonia Fluctuans
Two disorders that mimic myotonia congenita and are forms of sodium channel myotonia without periodic paralysis include acetazolamide-responsive sodium channel myotonia and myotonia fluctuans [Heatwole and Moxley, 2007] (Table 96-3).
Table 96-3 Sodium Channel Myotonias without Periodic Paralysis
| Clinical Features | Acetazolamide-Responsive Sodium Channel Myotonia | Myotonia Fluctuans |
|---|---|---|
| Inheritance | Dominant | Dominant |
| Gene defect | Chromosome 17; mutation in skeletal muscle sodium channel | Chromosome 17; mutation in skeletal muscle sodium channel |
| Age of onset | First decade | First or second decade |
| Myopathy | Rare | Rare, muscle hypertrophy common |
| Myotonia | Face, paraspinal muscles, paradoxical myotonia of eyelids, grip limbs; varies in severity and often there is pain with myotonia | Face, limbs, eyelids; frequently fluctuates in severity; especially after exercise |
| Provocative stimuli | Fasting, cold, oral potassium, infection | Exercise-rest-exercise, oral potassium |
| Therapy for symptoms | Acetazolamide, mexiletine; avoid high-potassium diet; monitor during and after surgery for rigidity and rhabdomyolysis | Mexiletine; avoid high-potassium diet; monitor during and after surgery for rigidity and rhabdomyolysis |
The discovery that hyperkalemic periodic paralysis and paramyotonia congenita result from mutations in the gene for the skeletal muscle sodium channel [Cannon, 2002; Lehmann-Horn and Jurkat-Rott, 1999; Jurkat-Rott and Lehmann-Horn, 2007] has led to further investigations that have identified a group of patients with myotonia without episodic weakness, whose myotonia is worsened by potassium intake and who have mutations in the gene for the skeletal muscle sodium channel [Lerche et al., 1993; Ptacek et al., 1994a; Ricker et al., 1990, 1994b; Heatwole and Moxley, 2007; Colding-Jorgensen et al., 2006; Gay et al., 2008; Kubota et al., 2009]. This section reviews two of these disorders, acetazolamide-responsive myotonia [Ptacek et al., 1994b] and myotonia fluctuans [Ricker et al., 1990, 1994b], which resemble chloride channel myotonia. A review of two other rare nondystrophic myotonic disorders caused by mutations in the sodium channel, myotonia permanens and chronic myotonia [Lerche et al., 1993; Colding-Jorgensen et al. 2006], is not included. The reader is referred to the reports describing these uncommon disorders.
Clinical Features
Acetazolamide-responsive sodium channel myotonia often presents initially to the pediatrician rather than the neurologist with common complaints, such as clumsiness causing frequent falls, a lazy eye, growing pains, back muscle spasm, or stridor. Parents may observe that, after prolonged crying, an infant or young child will have “their eyes stuck.” This symptom is a manifestation of paradoxical eyelid myotonia that develops with repeated bouts of forceful closure of the eyes during crying. Myotonia is most apparent in the face, eyes, and larynx in very young patients. Stiffness in the hands, proximal limb muscles, and paraspinous muscles is more apparent in middle childhood. Muscle hypertrophy, especially in the legs, is frequent. Cerebellar function, sensory testing, tendon reflexes, and muscle strength are normal. Grip and percussion myotonia, as well as lid lag, are present. The severity of these complaints varies both in affected individuals within the same kindred and between kindreds. Episodes of painful muscle spasm occur in some patients, usually during or immediately after exercise. Pain is relatively infrequent in recessive and dominant chloride channel myotonias [Trip et al., 2009a – see section above on chloride channel myotonia]. In contrast, pain is common in acetazolamide-responsive sodium channel myotonia and DM2.
The clinical findings in myotonia fluctuans are similar to those described previously for acetazolamide-responsive sodium channel myotonia, with one important additional finding. The severity of the muscle stiffness fluctuates to a greater degree in this illness. On “good days,” the myotonia can vanish. The fluctuating myotonia also has an interesting relationship to prolonged exercise. A phenomenon termed exercise-induced delayed-onset myotonia occurs [Ricker et al., 1990]. Patients report that, after a period of rest following vigorous exercise, if they resume exercise “at the wrong time,” severe muscle stiffness may develop. The time interval between the period of rest and the resumption of exercise is critical. If it is only a few minutes or an hour or more, stiffness does not occur. During the intermediate time interval, the fluctuation and provocation of severe myotonia happen.
Genetics
Acetazolamide-responsive sodium channel myotonia results from a mutation in the gene for the skeletal muscle sodium channel [Ptacek et al., 1994b], as does myotonia fluctuans [Ricker et al., 1994b]. The mutations in these two disorders of the sodium channel are not identical. Separate mutations produce myotonia fluctuans [Ricker et al., 1994b]. Both disorders are autosomal-dominant.
Pathophysiology
Both of these nondystrophic sodium channel myotonic disorders worsen with an elevation in extracellular potassium. The myotonic stiffness can disable a patient totally under these circumstances. The specific mechanism by which the fluctuation in severity of myotonia occurs remains unclear, but it may relate in part to hormonal fluctuations. The levels of insulin, corticosteroids, and other hormones may exert an important influence on extracellular potassium levels [Ricker et al., 1994b].
Clinical Laboratory Tests
Routine DNA testing is not available; however, several research laboratories screen for known mutations in the gene for the sodium channel in selected patients [Trip et al., 2009b; Fialho et al., 2007; Fournier et al., 2006]. The remainder of the laboratory evaluation is as described previously for chloride channel myotonia.
Treatment
Acetazolamide often controls myotonic stiffness and pain in acetazolamide-responsive sodium channel myotonia [Heatwole and Moxley, 2007; Ptacek et al., 1994b]. This treatment usually ameliorates the muscle pain. Annual ultrasound of the abdomen is useful to detect early formation of kidney stones, which are a common complication of acetazolamide [Tawil et al., 1993]. Dysesthesia and dysgeusia may also occur with this medication. For attacks of severe muscle spasm and pain, cyclobenzaprine can be tried. Patients undergoing surgery require monitoring during and after the procedure for signs of worsening muscle stiffness. Maintaining plasma potassium levels between 3.8 and 4.2 mEq/L helps to decrease perioperative muscle stiffness. Stress, muscle tissue damage, and bleeding all elevate plasma potassium levels and worsen myotonia. Postoperatively, intravenous diazepam or lorazepam may be necessary to decrease myotonic stiffness, especially if patients are unable to take medications by mouth. Mexiletine is an effective alternative to acetazolamide and, if necessary, these drugs can be used in combination [Heatwole and Moxley, 2007].
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


