Chapter 15 Chorea, ballism, and athetosis
Introduction
Chorea consists of involuntary, continual, abrupt, rapid, brief, unsustained, irregular movements that flow randomly from one body part to another. Patients can partially and temporarily suppress the chorea and frequently camouflage some of the movements by incorporating them into semipurposeful activities (parakinesia). The inability to maintain voluntary contraction (motor impersistence), such as manual grip (milkmaid grip) or tongue protrusion, is a characteristic feature of chorea and results in dropping of objects and clumsiness. Chorea should be differentiated from “pseudochoreoathetosis,” a movement disorder that is phenomenologically similar to chorea or athetosis (slow chorea) due to loss of proprioception (Sharp et al., 1994). Muscle stretch reflexes are often “hung-up” and “pendular.” Affected patients typically have a peculiar, irregular, and dance-like gait. The pathophysiology of chorea is poorly understood, but in contrast to parkinsonism, dystonia, and other movement disorders, intracortical inhibition of the motor cortex is normal in chorea (Hanajima et al., 1999). In addition, semiquantitative analysis of single photon emission computed tomography in patients with hemichorea due to various causes suggests that there is an increase in activity in the contralateral thalamus, possibly due to disinhibition as a result of loss of normal pallidal inhibitory input (Kim et al., 2002).
Chorea may be a manifestation of a primary neurologic genetic disorder, such as Huntington disease (HD), or it may occur as a neurologic complication of systemic, toxic, or other disorders (Rosenblatt et al., 1998; Cardoso, 2004; Cardoso et al., 2006; Jankovic and Fahn, 2009) (Tables 15.1 and 15.2, Fig. 15.1). Chorea may be seen in normal infants, but these movements usually disappear by age 8 months, and some of these movements may be purposeful (Van der Meer et al., 1995).
Table 15.1 Differential diagnosis of chorea
| Developmental choreas |
| Idiopathic choreas |
| Hereditary choreas |
Other heredodegenerations: Huntington disease-like (HDL) disorders (e.g., prion protein PRNP, junctophilin or JPH3 genes), dentatorubral-pallidoluysian atrophy (c-Jun NH-terminal kinase, JNK gene), spinocerebellar ataxias (SCA2, SCA17), ataxia telangiectasia, ataxia with oculomotor apraxia type 1 (aprataxin gene), ataxia with oculomotor apraxia type 2 (due to mutations in the senataxin gene), tuberous sclerosis of basal ganglia, pantothenate kinase associated neurodegeneration, other neurodegenerations with brain iron accumulation (Hallervorden–Spatz disease), Wilson disease, neuroferritinopathy, infantile bilateral striatal necrosis (IBSN) |
| Neurometabolic disorders |
| Drugs |
| Toxins |
| Metabolic and endocrine disorders |
| Infectious and postinfectious |
| Immunological |
| Vascular |
| Tumors |
| Trauma |
| Other secondary choreas |
| Miscellaneous |
Table 15.2 Differential diagnosis of inherited and sporadic choreas
| Inherited disorders | Sporadic disorders |
|---|---|
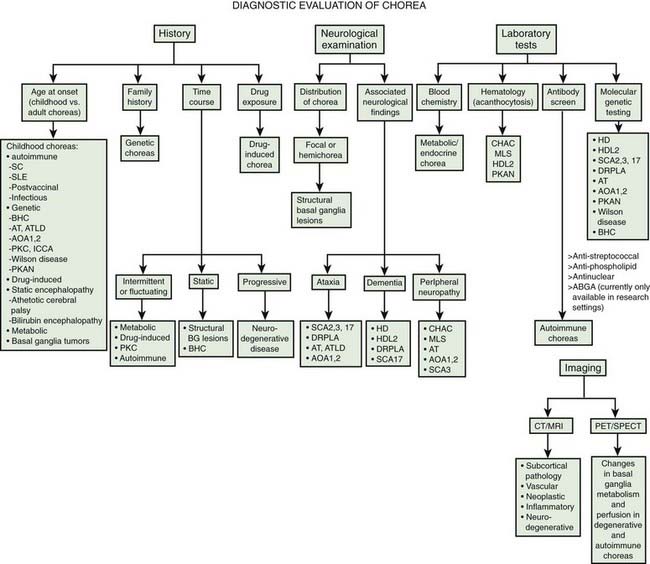
In this chapter we will briefly mention HD, but the focus will be on non-HD causes of chorea, as HD-like phenotypes without the HD genotype are increasingly being described and must be recognized. Several neurodegenerative disorders, some with expanded trinucleotide repeats, have been reported as phenocopies of HD, including spinocerebellar atrophy, particularly SCA2 and SCA3 (Kawaguchi et al., 1994), pure cerebello-olivary degeneration (Fox et al., 2003), and dentatorubral-pallidoluysian atrophy (DRPLA) (see below) (La Spada et al., 1994; Ikeuchi et al., 1995; Komure et al., 1995; Warner et al., 1995; Ross et al., 1997; Rosenblatt et al., 1998; Wild et al., 2008). In a study of 285 patients with clinical features consistent with HD, but who tested negative for HD by a DNA analysis, the following diagnoses were identified: five cases had Huntington disease-like type 4 (HDL4), one had HDL1, one had HDL2, and one patient had Friedreich ataxia (Wild et al., 2008).
Dentatorubral-pallidoluysian atrophy and HD-like disorders
Dentatorubral-pallidoluysian atrophy (DRPLA) is an autosomal dominant neurodegenerative disorder that is particularly prevalent in Japan, but it has been also identified in Europe and in African-American families (“Haw River syndrome”) (Burke et al., 1994; Thomas and Jankovic, 2001; Wardle et al., 2009) (Table 15.3). Usually beginning in the fourth decade, the disorder may occur as an early-onset DRPLA (before 20 years of age), manifested by a variable combination of myoclonus, epilepsy, and mental retardation, or late-onset DRPLA (after 20 years of age), manifested by cerebellar ataxia, choreoathetosis, dystonia, rest and postural tremor, parkinsonism, and dementia (Video 15.1). 
Table 15.3 Clinical features of dentatorubral-pallidoluysion atrophy (DRPLA)
| Early onset | Late onset |
|---|---|
Unstable CAG expansion has been identified as the mutation in the DRPLA (or ATN1) gene on chromosome 12p13.31 (Koide et al., 1994; Komure et al., 1995; Warner et al., 1995; Becher et al., 1997; Ross et al., 1997). The DRPLA gene codes for protein that has been identified as a phosphoprotein, c-Jun NH(2)-terminal kinase, one of the major factors involved in its phosphorylation. In DRPLA, this protein appears to be slowly phosphorylated; thus, it may delay a process that is essential in keeping neurons alive (Okamura-Oho et al., 2003). Similar to HD, there is an inverse correlation between the age at onset and the number of CAG repeats (Ikeuchi et al., 1995). The early onset of DRPLA is associated with greater number of CAG repeats (62–79) as compared to the late-onset type (54–67 repeats) (Ikeuchi et al., 1995). Testing for the various gene mutations will undoubtedly lead to better recognition and appreciation of the spectrum of clinical and pathologic changes associated with these disorders. For example, a family with spastic paraplegia, truncal ataxia, and dysarthria, but without other clinical features of DRPLA, has been found to show homozygosity for an allele that carries intermediate CAG repeats in the DRPLA gene (Kuroharas et al., 1997). The DRPLA gene is expressed predominantly in neurons, but neurons that are vulnerable to degeneration in DRPLA do not selectively express the gene (Nishiyama et al., 1997).
Neuroimaging studies in patients with DRPLA often show evidence of cortical, brainstem, and cerebellar atrophy and widespread white matter changes (Koide et al., 1997; Muñoz et al., 1999, 2004). Neuropathologic findings consist chiefly of degeneration of the dentatorubral system, globus pallidus externa (GPe), subthalamic nucleus, and, to a lesser extent, striatum, substantia nigra, inferior olive, and thalamus (Warner et al., 1994) as well as demyelination and reactive astrogliosis in the cerebral white matter (Muñoz et al., 2004). Involvement of oligodendrocytes in autopsied brains and an increased number of affected glia, as well as larger expansions in CAG in these glia, in transgenic mice might explain the widespread demyelination (Yamada et al., 2002). Several pathologic reports have noted widespread deposition of lipofuscin. Similar to HD and other diseases associated with CAG repeat expansions, DRPLA has also been associated with the formation of perinuclear aggregates that can be prevented by the use of transglutaminase inhibitors such as cystamine and monodansyl cadaverine (Igarashi et al., 1998). These intranuclear inclusions stain intensely with ubiquitin (Becher and Ross, 1998). Subsequent studies have demonstrated accumulation of mutant atrophin-1 in the neuronal nuclei, rather than neuronal intranuclear inclusions, as the predominant pathologic feature in this neurodegenerative disorder (Yamada et al., 2001).
Other Huntington disease-like disorders
An autosomal dominant HD-like neurodegenerative disorder, now classified as HDL1 and mapped to chromosome 20p (Xiang et al., 1998), is a familial prion disease with an expanded PrP. A 192-nucleotide insertion in the region of the prion protein gene (PRNP) encoding an octapeptide repeat in the prion protein, was found in a single family with HD phenotype, suggesting that PRNP mutations can result in HD phenocopies (Moore et al., 2001).
Another disorder, termed HDL2, is characterized by onset in the fourth decade, involuntary movements such as chorea and dystonia as well as other movement disorders (bradykinesia, rigidity, tremor), dysarthria, hyperreflexia, gait abnormality, psychiatric symptoms, weight loss, and dementia with progression from onset to death in about 20 years (Margolis et al., 2001, 2004; Walker et al., 2003a). The disorder appears to be present exclusively or predominantly in individuals of African origin. The neuroimaging and neuropathologic findings are very similar to those in HD, except that there appears to be more severe involvement of the occipital cortex (Greenstein et al., 2007), and the intranuclear inclusions stain with 1C2 but not with anti-huntingtin antibodies. Unlike the family linked to chromosome 20p, seizures are not present in the HDL2 family. All 10 affected family members had a CAG repeat expansion of 50–60 triplets. The gene was later mapped to chromosome 16q24.3 and was found to encode junctophilin-3, a protein of the junctional complex linking the plasma membrane and the endoplasmic reticulum (Holmes et al., 2001). Although acanthocytosis was emphasized by Walker and colleagues (2002, 2003b) in their initial report and in one of three patients reported subsequently (Walker et al., 2003b), we have not been able to confirm the presence of acanthocytes in one member of the original family or in the other members when we carefully examined the peripheral smear.
The mutation associated with HDL2 has been identified as a CTG/CAG trinucleotide repeat expansion within the junctophilin-3 (JPH3) gene. In the normal population, the repeat length ranges from 6 to 27 CTG triplets, whereas affected individuals have 41–58 triplets. One family, previously described as “autosomal dominant chorea–acanthocytosis with polyglutamine-containing neuronal inclusions” (see later) (Walker et al., 2002), was subsequently found to have the triple nucleotide expansion of HDL2 (Stevanin et al., 2003; Walker et al., 2003b). The CTG repeat expansion at the HDL2 locus has been found to be responsible for 2% of patients with typical features of HD but without expanded CAG repeats in the IT15 gene and 0.2% of all HD families, again providing evidence that HD is clinically and genetically heterogeneous (Stevanin et al., 2002). This group later analyzed 252 patients with an HDL phenotype, including 60 with typical HD, who had tested negative for pathologic expansion in the IT15 gene and found two patients that had an abnormal CTG expansion in the JPH3 gene and two other patients with abnormal CAG expansion in the gene coding for TATA-binding protein (TBP/SCA17), important in initiation of transcription (Stevanin et al., 2003; Toyoshima et al., 2004; Schneider et al., 2006). SCA17, categorized as HDL4, has many clinical features that overlap with HD (Schneider et al., 2007; Bech et al., 2010). Thus, the frequency of mutation in either the JPH3 gene or TBP gene among patients with HDL phenotype is about 3%. Initially, the TBP expansion was found in the family with SCA17, characterized clinically by intellectual deterioration, cerebellar ataxia, epilepsy, and chorea. CUG repeat-containing RNA foci, resembling myotonic dystrophy type 1, were detected in neurons of HDL2 brains, suggesting that RNA toxicity may play a role in the pathogenesis of this neurodegenerative disorder (Rudnicki et al., 2007). HLD2 resembles HD clinically and pathologically more than any other disease (Rudnicki et al., 2008).
While the majority of genetic forms of chorea are inherited in an autosomal dominant pattern, a novel autosomal recessive neurodegenerative HDL disorder has been described (Kambouris et al., 2000). Beginning at 3–4 years of age and manifested by chorea, dystonia, ataxia, gait disorder, spasticity, seizures, mutism, intellectual impairment, and bilateral frontal and caudate atrophy, this neurodegenerative disorder has been linked to 4p15.3, different from the 4p16.3 HD locus, but confirmation of this finding is lacking. Although classified as HDL3, because of its AR inheritance and clinical features atypical for HD, it should not be categorized as HDL. In fact the HDL terminology should be replaced with specific disorders in which genetic mutation has been identified.
Some cases of neuronal intranuclear inclusion disease, caused by expanded CAG repeats and characterized by the combination of extrapyramidal signs, lower motor neuron signs, and cognitive and behavioral abnormalities resulting in death by the third decade, also show intranuclear aggregates, similar to the other CAG disorders (Lieberman et al., 1998).
Neuroacanthocytosis
After HD, neuroacanthocytosis is perhaps the most common form of hereditary chorea (Table 15.4). Previously also referred to as chorea–acanthocytosis, it is now recognized that this multisystem, neurodegenerative disorder can be expressed by a wide variety of clinical and laboratory abnormalities, hence the term neuroacanthocytosis (Spitz et al., 1985; Danek et al., 2005; Walker et al., 2006, 2007; Thomas and Jankovic, 2006) (Table 15.5). The term was coined by Jankovic et al. (1985) to replace the old term Levine–Critchley syndrome choreoacanthocytosis to draw attention to the heterogeneous presentation with a variety of hyperkinetic (chorea, dystonia, tics) and hypokinetic (parkinsonism) movement disorders in addition to other neurologic deficits and abnormal laboratory findings. Symptoms usually first begin in the third and fourth decades of life (range: 8–62 years) with lip and tongue biting followed by orolingual (eating) dystonia, motor and phonic tics, generalized chorea, and stereotypies (Video 15.2). Other features include cognitive and personality changes, seizures, dysphagia, dysarthria, vertical ophthalmoparesis, parkinsonism, amyotrophy, areflexia, evidence of axonal neuropathy, and elevated serum creatine kinase without evidence of myopathy. Hardie and colleagues (1991) reviewed the clinical, hematologic, and pathologic findings in 19 patients (10 males and 9 females) with a mean age of 32 years (range: 8–62 years) with more than 3% acanthocytes on peripheral blood smear. Twelve of these patients with neuroacanthocytosis were familial, and seven were sporadic; two had the McLeod phenotype (see later). In their series, Hardie and colleagues (1991) found a variety of movement disorders, including chorea (58%), orofacial dyskinesia (53%), dystonia (47%), vocalizations (47%), tics (42%), and parkinsonism (34%). Although lip and tongue biting was observed in only 16% of the patients, this is a characteristic feature of neuroacanthocytosis and when present, it strongly suggests the diagnosis. The use of a mouth guard has been reported to be effective in the treatment of oral self-mutilation associated with neuroacanthocytosis (Fontenelle and Leite, 2008). Besides movement disorders other associated features included dysarthria (74%); absent or reduced reflexes (68%); dementia (63%); psychiatric problems such as depression, anxiety, and obsessive-compulsive disorder (58%); dysphagia (47%); seizures (42%); muscle weakness and wasting (16%); and elevated creatine phosphokinase (CK) in 58%. Magnetic resonance volumetry and fluorodeoxyglucose positron emission tomography (PET) show striatal atrophy in patients with neuroacanthocytosis (Jung et al., 2001). 
Table 15.4 Clinical and genetic features of neuroacanthocytosis
Table 15.5 Classification of neuroacanthocytosis
| I. Normal lipids |
| II. Hypobetalipoproteinemia (Mars et al., 1969) |
| III. Abetalipoproteinemia (Bassen and Kornzweig, 1950) |
| IV. Aprebetalipoproteinemia (Bohlega et al., 1998) |
| V. Hypoprebetalipoproteinemia |
| VI. X-linked (McLeod syndrome) (Allen et al., 1961) Xp21 |
Although autosomal dominant, X-linked recessive, and sporadic forms of neuroacanthocytosis have been reported, the majority of the reported families indicate autosomal recessive inheritance. Genome-wide scan for linkage in 11 families with autosomal recessive inheritance showed a linkage to a marker on chromosome 9q21, indicating a single locus for the disease (Rubio et al., 1997). Ueno and colleagues (2001) carried out a linkage-free analysis in the region of chromosome 9q21 in the Japanese population and identified a 260 bp deletion in the EST (expressed sequence tags) region K1AA0986 in exon 60, 61 that was homozygous in patients with neuroacanthocytosis and heterozygous in their parents. Further sequencing has identified a polyadenylation site with a protein with 3096 amino acid residues that has been named “chorein” by the authors. This deletion is not found in normal Japanese and European populations (Ueno et al., 2001). In another study by Rampoldi and colleagues (2001) in European patients, a novel gene encoding a 3174-amino-acid protein on chromosome 9q21 with 73 exons was identified. They identified 16 mutations in the chorea acanthocytosis (ChAc) gene, later renamed VPS13A gene. These mutations were identified in various exons. They suggested that chorea acanthocytosis encodes an evolutionarily conserved protein that is involved in protein sorting (Rampoldi et al., 2002). Other single heterozygous mutations have been identified in this gene (Saiki et al., 2003). Molecular analysis by screening all 73 exons of the VPS13A gene showed marked genotype–phenotype heterogeneity (Lossos et al., 2005). The function of the protein product, chorein, is not yet fully understood, but it is probably involved in intracellular protein trafficking. Using antichorein antisera, the expression of chorein in peripheral red blood cells has been found to be absent or markedly reduced in patients with neuroacanthocytosis, but not with McLeod syndrome or with HD (Dobson-Stone et al., 2004). Loss of chorein expression, measured by Western blot analysis has been found to be a reliable diagnostic test for neuroacanthocytosis.
Walker and colleagues (2002) described a family with chorea or parkinsonism as well as cognitive changes, inherited in an autosomal dominant pattern. At autopsy, there was marked degeneration of the striatum and intranuclear inclusion bodies immunoreactive for ubiquitin, expanded polyglutamine CGG repeats, and torsinA. Interestingly, one of the patients had fragile X syndrome, and two had expanded trinucleotide repeats at permutation range, previously associated with postural/kinetic tremor, parkinsonism, ataxia, and cognitive decline (Hagerman et al., 2001). The family reported by Walker and colleagues (2002) turned out to have the trinucleotide repeat expansion associated with HDL2, but subsequent analysis of the family shed doubt on the presence of acanthocytes as a feature of the HDL2 syndrome (Walker et al., 2003a).
Two patients from the original study by Rubio and colleagues (1997) were found to have the McLeod phenotype, an X-linked (Xp21) recessive form of acanthocytosis associated with depression, bipolar and personality disorder, and neurologic manifestations, including chorea, involuntary vocalizations, seizures, motor axonopathy, hemolysis, liver disease, and high creatine kinase levels (Witt et al., 1992; Danek et al., 2001a, 2001b). Neuroimaging usually reveals caudate and occasionally cerebellar atrophy with a rim of increased T2-intensity in the lateral putamen. Functional neuroimaging studies show evidence of downregulation of D2 dopamine receptors. In contrast to the autosomal recessive form of neuroacanthocytosis linked to mutations in VPS13A gene on chromosome 9, patients with McLeod syndrome usually do not exhibit lip-biting or dysphagia. This multisystem disorder is associated with low reactivity of Kell erythrocyte antigens (weak antigenicity of red cells) due to absence of XK, a 37 kDa, 444-amino-acid, membrane protein that forms a complex with the Kell protein. The disorder is caused by different mutations in the XK gene encoding for the XK protein (Ho et al., 1996; Danek et al., 2001a; Jung et al., 2001). Mutations identified by various authors include frame shift mutations in exon 2 at codon 151, deletion at codon 90 in exon 2 and at codon 408 in exon 3, and splicing mutations in intron 2 of the XK gene (Dotti et al., 2000; Ueyama et al., 2000; Danek et al., 2001a; Jung et al., 2001). Rarely, neuroacanthocytosis may be associated with abetaliproteinemia due to mutations in the microsomal triglyceride transfer protein (Sharp et al., 1993). In addition to acanthocytosis, the patients exhibit retinopathy; malabsorption, including that of vitamin E; low serum cholesterol levels; and abnormal serum lipoprotein electrophoresis. Aprebetalipoproteinemia can also cause movement disorders and acanthocytosis (Bohlega et al., 1998).
An examination of wet blood or Wright-stained, fast dry, blood smear usually reveals over 15% of red blood cells as acanthocytes. In mild forms of acanthocytosis, scanning electron microscopy might be required to demonstrate the red blood cell abnormalities (Feinberg et al., 1991). In a recent study of two patients with pathologically proven neuroacanthocytosis, Feinberg and colleagues (1991) noted that the yield in demonstrating acanthocytosis may be increased by using a coverslip because the contact with glass causes the fragile cells to undergo morphologic changes. Diluting the blood with normal saline, incubating the Wright-stained smear with EDTA, using a scanning electron microscope, and other techniques designed to increase “echinocytotic stress” are also helpful (Feinberg et al., 1991; Orrell et al., 1995). The characteristic acanthocytic appearance of red blood cells has been attributed to abnormalities in transmembrane glycoprotein band 3 that can be demonstrated on gel electrophoresis. It is not yet clear how the gene mutation leads to the abnormal morphology of the red cells.
By using high-performance liquid chromatography, fatty acids of erythrocyte membrane proteins were analyzed in six patients with neuroacanthocytosis (Sakai et al., 1991). In comparison with normal controls and patients with HD, erythrocytes of patients with neuroacanthocytosis showed a marked abnormality in the composition of covalently bound fatty acids: an increase in palmitic acid (C16:0) and a decrease in stearic acid (C18:0).
Brain magnetic resonance imaging (MRI) in patients with neuroacanthocytosis usually shows caudate and more generalized brain atrophy, but some cases also show extensive white matter abnormalities (Nicholl et al., 2004). Caudate hypometabolism and atrophy have been demonstrated by PET studies and by neuroimaging. Similar to the findings in Parkinson disease, PET scans in six patients with neuroacanthocytosis showed a reduction to 42% of normal in [18F]dopa uptake in the posterior putamen; in contrast to Parkinson disease, however, there was a marked reduction in the striatal [11C]raclopride (D2) receptor binding (Brooks et al., 1991).
Neuronal loss and gliosis were particularly prominent in the striatum and pallidum but may also affect the thalamus, substantia nigra, and anterior horns of the spinal cord (Rinne et al., 1994b). The neuronal loss in the substantia nigra is most evident in the ventrolateral region, similar to Parkinson disease, but the nigral neuronal loss is more widespread in neuroacanthocytosis (Rinne et al., 1994a). The preservation of the cerebral cortex, cerebellum, subthalamic nucleus, pons, and medulla may serve to differentiate pathologically between neuroacanthocytosis, HD, and DRPLA. Brain biochemical analyses showed low substance P in the substantia nigra and striatum and increased levels of norepinephrine in the putamen and pallidum (DeYebenes et al., 1988).
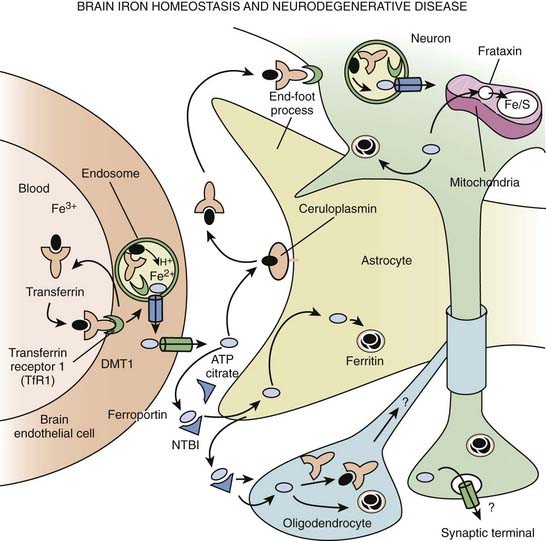
Neurodegeneration with brain iron accumulation
A group of neurodegenerative disorders, formerly known as Hallervorden–Spatz disease, has been receiving increasing attention as the genetic and pathogenic mechanisms of the various subtypes become elucidated. Because of Hallervorden’s terrible past and his shameless involvement in active euthanasia (Shevell, 2003), this group of disorders has been renamed neurodegeneration with brain iron accumulation (NBIA). The prototype form of NBIA consists of an autosomal recessive disorder characterized by childhood-onset progressive rigidity, dystonia, choreoathetosis, spasticity, optic nerve atrophy, and dementia, and has been associated with acanthocytosis (Malandrini et al., 1996; Racette et al., 2001; Thomas et al., 2004). Although chorea is not a typical feature of NBIA, “senile chorea” has been described in a patient with pathologically proven NBIA type1 (NBIA-1) (Grimes et al., 2000).
The most classic NBIA, NBIA-1, is the pantothenate kinase-associated neurodegeneration (PKAN). Linkage analyses initially localized the NBIA-1 gene on 20p12.3–p13; subsequently, a 7 bp deletion and various missense mutations were identified in the coding sequence of the PANK2 gene, which codes for pantothenate kinase (Zhou et al., 2001; Hayflick et al., 2003). Pantothenate kinase is an essential regulatory enzyme in coenzyme A biosynthesis. It has been postulated that as a result of phosphopantothenate deficiency, cysteine accumulates in the globus pallidus of brains of patients with NBIA-1. It undergoes rapid auto-oxidation in the presence of nonheme iron that normally accumulates in the globus pallidus interna (GPi) and substantia nigra, generating free radicals that are locally neurotoxic. Interestingly, atypical subjects were found to be compound heterozygotes for certain mutations for which classic subjects were homozygous. The disorder with the clinical phenotype of NBIA associated with mutations in the PANK2 gene is now referred to as pantothenate kinase-associated neurodegeneration or PKAN (Thomas et al., 2004). On the basis of an analysis of 123 patients from 98 families with NBIA-1, Hayflick and colleagues (2003) found that “classic Hallervorden–Spatz syndrome” was associated with PANK2 mutation in all cases and that one-third of “atypical” cases had the mutations within the PANK2 gene. Those who had the PANK2 mutation were more likely to have dysarthria and psychiatric symptoms, and all had the typical “eye-of-the-tiger” abnormality on MRI with a specific pattern of hyperintensity within the hypointense GPi (McNeill et al., 2008).
Neuroacanthocytosis and NBIA may overlap in some clinical features. While PKAN may be associated with acanthocytosis, another neuroacanthocytosis syndrome, linked to PKAN, is the hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration (HARP) syndrome (Orrell et al., 1995). This disorder is associated with dystonia, particularly involving the oromandibular region, rather than chorea and self-mutilation. Indeed, a homozygous nonsense mutation in exon 5 of the PANK2 gene that creates a stop codon at amino acid 371, found in the original HARP patient, establishes that HARP is part of the pantothenate kinase-associated neurodegeneration disease spectrum (Ching et al., 2002; Houlden et al., 2003).
The classification of NBIA is continuously being revised as our understanding of this group of disorders is improving. In addition to PKAN, other forms of NBIA include neuroferritinopathy, infantile neuroaxonal dystrophy, and aceruloplasminemia, and PLA2G6-associated neurodegeneration (PLAN), with mutations in the PLA2G6 gene, on chromosome 22q13.1 (Schneider et al., 2009) (Table 15.6, Fig. 15.2). These disorders are chiefly manifested by childhood-onset axial hypotonia, spasticity, bulbar dysfunction, ataxia, dystonia, and choreoathetosis, as well as MRI changes indicative of iron deposition in the globus pallidus and substantia nigra (Kurian et al., 2008). Previously diagnosed as infantile neuroaxonal dystrophy, now classified as NBIA-2, PLAN may also present as adult-onset, levodopa-responsive dystonia–parkinsonism without iron on brain imaging (McNeill et al., 2008; Paisan-Ruiz et al., 2008; Schneider et al., 2009). Another gene, FA2H, when mutated, has been found to cause not only leukodystrophy and hereditary spastic paraplegia, but also NBIA (Kruer et al., 2010). The FA2H-associated neurodegeneration (FAHN) is characterized by childhood-onset gait impairment, spastic paraparesis, ataxia, and dystonia (Kruer et al., 2010; Schneider and Bhatia, 2010). FA2H is involved in lipid and ceramide metabolism. Another form of NBIA was highlighted by a report of 11 children with a biochemical profile suggestive of dopamine transporter deficiency syndrome (Kurian et al., 2011). Presenting in infancy, this disorder is usually characterized by severe parkinsonism-dystonia syndrome, but chorea, oculomotor deviations, and spasticity may also dominate the clinical phenotype. The CSF ratio of homovanillic acid to 5-hydroxyindoleacetic acid is usually increased. This autosomal recessive disorder has been attributed to homozygous or compound heterozygous SLC6A3 mutations and complete loss of dopamine transporter activity in the basal nuclei (indicated by abnormal DAT Scan SPECT). Regression of symptoms after 6 months of iron chelation with deferiprone has been reported in some patients with NBIA (Forni et al., 2008).
Table 15.6 Differential diagnosis of neurodegeneration with brain iron accumulation (NBIA)
Other familial choreas
Besides HD and neuroacanthocytosis, genetically transmitted choreas include benign hereditary chorea (BHC), a nonprogressive chorea of childhood onset (Wheeler et al., 1993; Kleiner-Fisman and Lang, 2007; Adam and Jankovic, 2010) (Table 15.7). BHC usually starts in early childhood and progresses until the second decade, after which time it remains static or even spontaneously improves. The patients may have a slight motor delay because of chorea, slight gait ataxia, and their handwriting may be impaired, but the disorder is self-limiting after adolescence in most cases, although it may persist as a mild chorea beyond age 60 years. Inherited as an autosomal dominant disorder, BHC has been linked to a marker on chromosome 14q13.1–q21.1 (de Vries et al., 2000; Fernandez et al., 2001; Breedveld et al., 2002) and a novel single nucleotide substitution in the TITF1 gene (also referred to as TTF, Nkx2.1, and T/ebp), coding for a transcription essential for the organogenesis of the lung, thyroid, and basal ganglia, has been identified in one Canadian family (Kleiner-Fisman et al., 2003; Kleiner-Fisman and Lang, 2007; Ferrara et al., 2008) (http://www.ncbi.nlm.nih.gov/sites/GeneTests/). This syndrome has been also described in patients with deletion of not only the TITF1 gene but also the contiguous PAX9 gene (Devos et al., 2006). These mutations should be considered in children and adults with chorea, mental retardation, congenital hypothyroidism, and chronic lung disease, hence the term brain–lung–thyroid syndrome proposed for this disease (Willemsen et al., 2005; Devos et al., 2006). MRI has been generally reported to be unremarkable but some cases showed hypoplastic pallidum, lack of differentiation of medial and lateral components, and bilateral signal hyperintensities on T2-weighted MRI images (Kleiner-Fisman and Lang, 2007). Two autopsied brains from patients with BHC due to TITF1 showed no pathologic abnormalities (Asmus et al., 2005), but specific immunochemical investigations suggested reduced number of striatal and neocortical interneurons, consistent with a defect in migration normally mediated by the TITF1 gene (Kleiner-Fisman et al., 2005).
Table 15.7 Clinical features of benign hereditary chorea
Adult-onset, autosomal dominant, benign chorea without dementia, initially reported in Japan, in which HD, HDL1, HDL2, DRPLA, SCA17, and mutations in the TITF1 gene were excluded, revealed linkage to a novel locus on chromosome 8q21.3–q23.3 in two Japanese families with “benign hereditary chorea type 2” (BHC2) (Shimohata et al., 2007). Surprisingly, BHC may improve markedly with levodopa (Asmus et al., 2005). The existence of BHC has been questioned because many patients who were initially diagnosed with the disorder were later found to have some other diagnosis, such as myoclonic dystonia, hereditary essential myoclonus, tics, and HD (Schrag et al., 2000).
Essential chorea is a form of adult-onset, nonprogressive chorea without family history of chorea or other symptoms suggestive of HD and without evidence of striatal atrophy on neuroimaging studies. Sometimes referred to as senile chorea, essential chorea usually has its onset after age 60, and in contrast to HD, it is not associated with dementia or positive family history. Some cases of senile chorea, however, have been reported to have pathologic changes identical to HD; others have had predominant degeneration of the putamen rather than the caudate (Friedman and Ambler, 1990). The CAG repeat length should, by definition, be normal, but Ruiz and colleagues (1997) found abnormal CAG expansion in three of six clinically diagnosed cases of senile chorea. Although the authors suggest that some patients with senile chorea have a sporadic form of HD, the term essential (or senile) chorea should be reserved for those patients with late-onset chorea without family history, without dementia, without psychiatric problems, and without CAG expansion. These criteria are necessary in order to separate senile or essential chorea from HD. Hereditary chorea without dementia and with a benign course has been also described (Behan and Bone, 1977).
Choreoathetosis, along with developmental regression, mental retardation, pendular nystagmus, optic atrophy, dysphagia, dystonia, spasticity, and severe bilateral striatal atrophy with response to biotin, is a feature of familial infantile bilateral striatal necrosis (IBSN) (Straussberg et al., 2002; Basel-Vanagaite et al., 2006). IBSN, an autosomal recessive neurodegenerative disorder, was found to be associated with mutation of nup62 on chromosome 19q13.32–13.41 (Basel-Vanagaite et al., 2006).
Ataxia telangiectasia, an autosomal recessive multisystem disease, is another disorder often associated with chorea. In a retrospective analysis of the clinical characteristics in 18 adult patients with ataxia telangiectasia from 9 families and 6 unrelated adults documented with elevated alpha-fetoprotein, chromosomal instability, and mutations in the A-T mutated (ATM) gene and measurements of ATM protein expression and kinase activity, chorea–athetosis was present in 14/18 (78%) cases (Verhagen et al., 2009). Other common abnormalities included dysarthria, oculomotor apraxia, dystonia, rest tremor, neuropathy, immunodeficiency, restricted respiratory function, and malignancies. Not all affected individuals have telangiectasia.
Another disorder, rarely considered in the differential diagnosis of chorea, is familial dyskinesia and facial myokymia (FDFM), characterized by childhood-onset adventitious movements and perioral or periorbital myokymia. These movements may be paroxysmal in early stages, increase in frequency and severity, and become constant in the third decade, without intellectual impairment or shortening of lifespan. Frontotemporal dementia, particularly associated with TAR-DNA binding protein (TDP)-43 abnormalities due to TARDBP mutations, may be associated not only with amyotrophic lateral sclerosis, but also supranuclear gaze palsy and chorea (Kovacs et al., 2009).
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


