Chromosomal Anomalies and Contiguous-Gene Syndromes
John H. Menkes
Rena E. Falk
This chapter considers conditions that develop as a result of chromosomal abnormalities. Abnormalities of chromosomes are either constitutional or acquired. Acquired chromosomal abnormalities develop postnatally, affect only one clone of cells, and are implicated in the evolution of neoplasia. Constitutional abnormalities, on the other hand, develop during gametogenesis or early embryogenesis and affect a significant portion of an individual’s cells. Until recently, detection of constitutional abnormalities was limited to disorders that resulted in gains or losses of an entire chromosome or large portions thereof—abnormalities readily visible by standard cytogenetics. The smallest loss or gain visible with standard cytogenetic preparations is about 4 megabases (Mb) of DNA (1). The introduction of chromosomal banding techniques with increasing degrees of resolution, the widespread use of fluorescence in situ hybridization (FISH), and supplemental molecular analyses have refined cytogenetic analysis and have permitted detection of uniparental disomies and innumerable subtle, often submicroscopic (cryptogenic), deletions, duplications, or translocations. This has removed any clear dividing line between changes considered to be chromosomal abnormalities and those considered to be molecular defects (1). Because in humans more than 30,000 genes are expressed in brain, it comes as no surprise that impaired brain function is the most common symptom of chromosomal anomalies.
In considering the significance of some of the subtle chromosomal abnormalities, one must keep in mind that many of the abnormal chromosomal constitutions have been ascertained by searching the genome of mentally or neurologically handicapped individuals with multiple congenital abnormalities, thus introducing an obvious sampling bias. To complicate matters, chromosomal abnormalities are seen in only a fraction of handicapped individuals, whereas a sizable proportion of healthy adults have been found to have chromosomal variants. These include variations in Y chromosomal length, variations in satellite size or location, and variations in size of secondary constrictions as well as some forms of mosaicism, balanced translocations, and differences in the intensity and appearance of bands revealed by staining techniques. Small supernumerary chromosomes (marker chromosomes) may serve as a case in point. This abnormality is seen in 0.3 to 1.2 of 1,000 newborns and in 0.7 to 1.5 of 1,000 fetuses on amniocentesis. Even though the origin of the marker chromosome may be ascertained by FISH, for the purpose of genetic counseling, the prognosis of the marker is still uncertain in many cases (2).
An evaluation of the relationship of chromosomal abnormalities to neurologic deficits can be achieved by controlled, blind-coded cytogenetic surveys in groups of unclassifiable individuals with mental retardation and control individuals. One such study published in 1977 and using routine cytogenetics detected chromosomal abnormalities in 6.2% of patients with mental retardation and 0.7% of controls (3). Using banding techniques, Jacobs and colleagues found a 0.92% prevalence of structural chromosomal abnormalities in unselected newborns (4). Small chromosomal rearrangements affecting the telomeres (the specialized structure at the tips of chromosomes) have been detected in some 5% of individuals with mental retardation and in up to 7.4% of individuals with moderate to severe mental retardation (5,6). This relatively high yield prompts the use of such techniques in the investigation of children with global developmental delay, even in the absence of dysmorphic features (7). However, the cost of a systematic surveillance of the newborn population using high-resolution chromosomal banding, DNA polymorphisms, and subtelomeric FISH panels remains prohibitive.
The limitations of prenatal and neonatal surveillance programs are discussed more fully by Hook and colleagues (8). Studies in other species, as well as in humans, have shown that major duplications or deficiencies in autosomes tend to be lethal (9), with death occurring either in utero or in the immediate postnatal period (10). Few of the autosomal trisomies are compatible with survival.
Thus, the most common trisomy, that of chromosome 16, accounts for 7.5% of all spontaneous abortions but is not compatible with live birth, and only 2.8% of trisomy 13, 5.4% of trisomy 18, and 23.8% of trisomy 21 cases are live born (8).
Thus, the most common trisomy, that of chromosome 16, accounts for 7.5% of all spontaneous abortions but is not compatible with live birth, and only 2.8% of trisomy 13, 5.4% of trisomy 18, and 23.8% of trisomy 21 cases are live born (8).
AUTOSOMAL ABNORMALITIES
Down Syndrome (Trisomy 21)
Down syndrome (trisomy 21), the most common autosomal anomaly in live births and the most common single cause of mental retardation, was first differentiated from other forms of mental retardation in 1866 by Down (15) and Seguin (16). Demographic factors, notably the higher proportion of older women bearing children, has caused an increase in the incidence of this condition. However, prenatal diagnosis programs also have affected the incidence. The most recent figures in the United States (1997) indicate an incidence of 9.9 per 10,000 live births, and 43% occur in births to women aged 35 years and older (17). This is significantly less than the incidence of 1 in 500 recorded in 1950, and reflects the effectiveness of prenatal screening programs (18).
Etiology and Pathogenesis
Down syndrome is associated with either an extra chromosome 21 or an effective trisomy for chromosome 21 by its translocation to another chromosome, usually chromosome 14, or, less often, to another acrocentric chromosome, especially chromosome 21 or 22 (19).
A few terms require definition. In the general population about 1 couple in 500 carries a reciprocal translocation. A reciprocal translocation is a structural alteration of chromosomes that usually results from breakage of two chromosomes with the subsequent exchange of the resultant chromosomal segments. In general, this event occurs most frequently at prophase of the first meiotic cell division. The segments exchanged are nearly always from nonhomologous chromosomes and are usually not the same size. Such an rearrangement is said to be balanced when none of the chromosomal material appears to be duplicated or deleted on standard or high-resolution analysis.
Robertsonian translocation results from centromeric fusion of two acrocentric chromosomes. A larger chromosome is formed, with the resultant loss of the short-arm regions of the participating chromosomes, resulting in a complement of 45 chromosomes in the balanced carrier. In a survey that was based on prenatal genetic studies performed with banding the frequency of balanced chromosomal rearrangements was 1 in 250 or 0.4%. Reciprocal translocations were seen in 0.17%, inversions in 0.12%, and Robertsonian translocations in 0.11% (19a). On a clinical basis, it is impossible to distinguish between patients with Down syndrome caused by ordinary trisomy 21 and those caused by translocation. The frequency of trisomy 21 increases with maternal age and reaches an incidence of 1 in 54 births in mothers aged 45 years or older (20). In contrast to ordinary trisomy, the incidence of translocation Down syndrome is independent of maternal age. Most translocations producing Down syndrome arise de novo in the affected child and are not associated with familial Down syndrome. Data obtained between 1989 and 1993 on prenatal and postnatal diagnoses of 1,811 Down syndrome children born to mothers aged 29 years or younger show regular trisomy 21 in 90.8% (19). Of a group of 235 translocation Down syndrome individuals, 42% of cases involved a translocation between chromosome 21 and chromosome 14, whereas 38% showed a translocation involving two 21 chromosomes. In translocation patients for whom cytogenetic results were available from both parents, 21;21 translocations were almost invariably de novo, whereas 37% of 14;21 translocations were inherited from the mother and 4% derived from the father (19). Of 51 cases of 21;21 translocation investigated, 50 were de novo and one was of maternal origin (19).
Approximately 1% of couples experience the recurrence of Down syndrome as a result of regular trisomy 21. In most instances, this is caused by previously unrecognized mosaicism in one parent (21). In families in whom Down syndrome results from a translocation, the risk for subsequent affected children is significantly higher; it is 15% if the mother is the carrier, but less than 5% if the father is the translocation carrier (22).
By the use of polymorphic DNA markers, several studies have concluded that the extra chromosome is maternally derived and mainly is the consequence of errors in the first meiotic division in more than 90% of cases (23). The mean maternal age associated with errors in meiosis is approximately 33 years, significantly higher than the mean reproductive age in the United States. In 4.3%, there were errors in paternal meiosis. In these cases, and in the remainder, caused by errors in mitosis, maternal age is not increased (24). Several explanations have been put forth to account for the maternal age effect. With increasing age there are a decreasing number of chiasmata, that is, crossovers, sites where recombination occurs because of breakage and reunion between homologous chromosomes (25). The most likely hypothesis is that the decrease in chiasmata contributes to nondisjunction by disrupting the process of separation of the chromosomal homologues.
To understand the molecular pathogenesis of Down syndrome, two questions must be answered: (a) How does
the presence of a small acrocentric chromosome in triplicate and the increased expression of genes on this chromosome produce the anatomic and functional abnormalities of the Down syndrome phenotype, and (b) are we able to identify specific regions of chromosome 21 whose duplication is responsible for each of the phenotypic features of Down syndrome?
the presence of a small acrocentric chromosome in triplicate and the increased expression of genes on this chromosome produce the anatomic and functional abnormalities of the Down syndrome phenotype, and (b) are we able to identify specific regions of chromosome 21 whose duplication is responsible for each of the phenotypic features of Down syndrome?
TABLE 4.1 Genes Localized to Chromosome 21 Believed to Affect Brain Development, Neuronal Loss, and Alzheimer’s Type Pathology | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||
The sequencing of chromosome 21 has resulted in the identification of more than 300 genes, of which at least 165 have been confirmed experimentally (26). Of these at least 9 are believed to affect brain development and function (Table 4.1). Studies on individuals with partial trisomy 21 have suggested that overexpression and subsequent interactions of genes on chromosome 21q22 contribute significantly to the phenotype of Down syndrome (25,27). Two genes in this region have received particular attention. The gene for a Down syndrome cell adhesion molecule (DSCAM) is expressed in the developing nervous system, and, like other cell adhesion molecules, this molecule is critical for cellular migration, axon guidance, and the formation of neural connections (see Chapter 5) (25). Another gene localized to the 21q22 region is that for DYRK1A, a serine/threonine protein kinase, required for postembryonic proliferation of neuronal cell types. The overexpression of this gene is believed to interfere with postembryonic neurogenesis (27). However, the 21q22 region can hardly be considered “critical” for the production of the clinical syndrome, and duplication of genes in regions distinct from 21q22 are believed to be responsible for many of the typical Down syndrome features (28).
It also is not known how the presence of extra copies of genes and their regulatory sequences produces the Down syndrome phenotype. Although, on a theoretical basis, one would expect gene product levels 1.5 times normal, this is not always the case. For example, expression of the amyloid precursor protein (APP) is approximately four times normal in the brains of patients with Down syndrome (29). Its role in the development of early-onset Alzheimer disease in patients with Down syndrome is considered in the subsequent section.
Pathology
Malformations affect principally the heart and the great vessels. Approximately 20% to 60% of Down syndrome individuals have congenital heart disease. Defects in the atrioventricular septum were the most common abnormalities encountered by Rowe and Uchida in 36% of their patients, most of whom were younger than 2 years of age (30). A ventricular septal defect was present in 33% and a patent ductus arteriosus was present in 10% of patients. Among the other malformations, gastrointestinal anomalies were the most common. These included duodenal stenosis or atresia, anal atresia, and megacolon. Hirschsprung disease was seen in approximately 6% of patients (31).
Malformations of the spine, particularly of the upper cervical region, can occasionally produce neurologic symptoms. These are considered at another point of this chapter.
Malformations of the spine, particularly of the upper cervical region, can occasionally produce neurologic symptoms. These are considered at another point of this chapter.
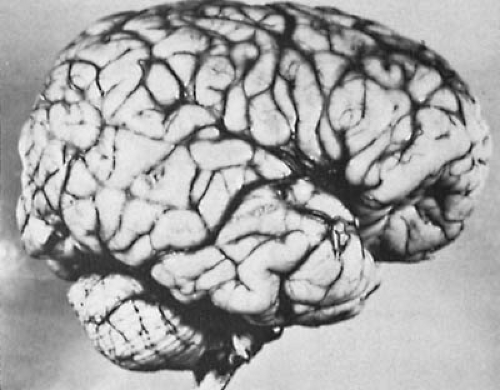 FIGURE 4.1. Lateral view of the brain of a 10-year-old boy with Down syndrome showing the typical small superior temporal gyrus and the large operculum. (From Lemire RJ, Loeser JD, Alvord C. Normal and abnormal development of the human nervous system. Hagerstown, MD: Harper & Row, 1975. With permission.) |
Grossly, the brain is small and spherical, and the frontal lobes, brainstem, and cerebellum are smaller than normal. The number of secondary sulci is generally reduced, the tuber flocculi persist, and the superior temporal gyrus is poorly developed (Fig. 4.1). A number of microscopic abnormalities also have been described (32,33). In essence, these involve reduction of neuronal density in various areas of the cortex, a loss of cortical interneurons, an accumulation of undifferentiated fetal cells in the cerebellum, and a reduction in the number of spines along the apical dendrites of pyramidal neurons, believed to indicate a reduction in the number of synaptic contacts (34). Interestingly, the dendritic tree is greater than normal in early infancy, but the excessive early outgrowth is followed by atrophy (35). Becker and colleagues explained these observations as an abortive attempt by neurons to compensate for the decreased number of spines and synapses on their receptive surfaces (35).
Abnormalities of the basal nucleus have received particular attention because an early and significant loss of cholinergic neurons in this area is a feature of both Down syndrome and Alzheimer disease. In Down syndrome the cell count is one-third normal, even before the onset of dementia, and it decreases progressively with age (36).
The most intriguing neuropathologic observation is the premature development of changes within the brain compatible with a morphologic diagnosis of Alzheimer disease. The spatial pattern of involvement of the brain by senile plaques and tangles follows that seen in Alzheimer disease. Thus, the amygdala, hippocampus, and the association areas of the frontal, temporal, and parietal cortex are particularly vulnerable to these changes (37). They are found in 15% of patients with Down syndrome aged 11 to 20 years, in 36% of patients aged 21 to 30 years, and in all patients who died aged 31 years or older (38). Microscopic alterations include pigmentary degeneration of neurons, senile plaques, and calcium deposits within the hippocampus, basal ganglia, and cerebellar folia. The calcium deposits in the cerebellar folia can be extensive enough to be seen on neuroimaging studies. As in Alzheimer disease, the brains of individuals with Down syndrome have reduced choline acetyltransferase activity, not only in areas that contain plaques, but also in those that appear microscopically normal (39,40).
Underlying the evolution of Alzheimer disease in children with Down syndrome is an excessive buildup of amyloid β-protein (AβP). This buildup is believed to be the consequence of an overexpression of the gene for amyloid β precursor protein (AβPP), mapped to chromosome 21q21. AβPP is cleaved to form AβP through the sequential action of beta-secretase and gamma-secretase. The gene for beta-secretase is also located on chromosome 21q22, and levels of this enzyme are significantly higher in Down syndrome fetal tissue than in controls, an indication that this gene is also overexpressed in Down syndrome (41). Cleavage of AβP by beta-secretase produces Aβ40, a soluble intracellular form, and plasma and tissue levels of soluble Aβ40 are higher in individuals with Down syndrome than in controls (42,43). Longer, less soluble forms, Aβ42 and Aβ43, are produced by the action of gamma-secretase, and Aβ42 is present in the brain of fetuses affected with Down syndrome as early as 21 weeks’ gestation (44,45). These insoluble aggregates of AβP readily form fibrils and are responsible for the ultimate appearance of both senile plaques and amyloid deposits in cerebral blood vessels (46). Why the appearance of plaques and amyloid deposits is delayed for decades after the appearance of Aβ42 remains unclear. Also unclear is the role played by mitochondrial dysfunction in the abnormal processing of AβPP (47).
Clinical Manifestations
The clinical picture of Down syndrome is protean and consists of an unusual combination of anomalies rather than a combination of unusual anomalies (22). The condition is seen more frequently in male individuals, with one study showing a male-to-female ratio of 1.23 to 1, but in mosaic individuals the male-to-female ratio is 0.8 to 1 (19).
The birth weight of Down syndrome infants is less than normal, and 20% weigh 2.5 kg or less (48). Neonatal complications are more common than normal, a consequence of fetal hypotonia and a high incidence of breech presentations. Physical growth is consistently delayed, and the
adult Down syndrome individual has a significantly short statue (49). The mental age that is ultimately achieved varies considerably. It is related in part to environmental factors, including the age at institutionalization for non–home-reared individuals, the degree of intellectual stimulation, and the evolution of presenile dementia even before puberty. As a rule, the developmental quotient (DQ) is higher than the IQ and decreases with age (Table 4.2) (50). Most older patients with Down syndrome have IQs between 25 and 49, with the average approximately 43. The social adjustment tends to be approximately 3 years ahead of that expected for the mental age (50). Del Bo and coworkers found that the decline in full-scale IQ with age was faster in those individuals who carry the apolipoprotein E4 allele, an important risk factor for both sporadic and familial late-onset Alzheimer disease (51).
adult Down syndrome individual has a significantly short statue (49). The mental age that is ultimately achieved varies considerably. It is related in part to environmental factors, including the age at institutionalization for non–home-reared individuals, the degree of intellectual stimulation, and the evolution of presenile dementia even before puberty. As a rule, the developmental quotient (DQ) is higher than the IQ and decreases with age (Table 4.2) (50). Most older patients with Down syndrome have IQs between 25 and 49, with the average approximately 43. The social adjustment tends to be approximately 3 years ahead of that expected for the mental age (50). Del Bo and coworkers found that the decline in full-scale IQ with age was faster in those individuals who carry the apolipoprotein E4 allele, an important risk factor for both sporadic and familial late-onset Alzheimer disease (51).
TABLE 4.2 Intellectual Functioning of Children with Down Syndrome | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||
Mosaicism can be found in approximately 2% to 3% of patients with Down syndrome. In these instances, the patient exhibits a mixture of trisomic and normal cell lines. As a group, these individuals tend to achieve somewhat higher intellectual development than nonmosaic individuals, and, in particular, they possess better verbal and visual-perceptual skills (52).
Aside from mental retardation, specific neurologic signs are rare and are limited to generalized muscular hypotonia and hyperextensibility of the joints. This is particularly evident during infancy and occurs to a significant degree in 44% of patients younger than 9 years of age (53).
Whereas major motor seizures are seen no more frequently in Down syndrome infants than in the general population, the association between infantile spasms and Down syndrome is greater than chance (54). Attacks can occur spontaneously or, less often, follow insults such as perinatal hypoxia. The electroencephalogram (EEG) demonstrates hypsarrhythmia or modified hypsarrhythmia (55). In the experience of Nabbout and coworkers a 6-month course of vigabatrin monotherapy has been effective, and with prompt treatment the prognosis for seizure control and preservation of intelligence is relatively good (54,56). In the experience of Stafstrom and Konkol, only 3 of 16 patients with Down syndrome with infantile spasms had persistent seizures, and the degree of cognitive impairment did not exceed that expected for the general Down syndrome population (55). A progressive disorganization of the EEG accompanies the evolution of Alzheimer dementia (57). Neuroimaging characteristically reveals large opercula, an indication of the underdeveloped superior temporal gyrus, and, in older patients, evidence exists of premature aging (58). Symmetric calcifications in basal ganglia, notably in the globus pallidus, are common, particularly in adults (59,60).
Numerous case reports and series have associated atlantoaxial instability and dislocation with Down syndrome. The instability, seen in some 15% to 40% of patients, results from laxity of the transverse ligaments that hold the odontoid process close to the anterior arch of the atlas (61). The instability is usually asymptomatic, but neck pain, hyperreflexia, progressive impairment of gait, and urinary retention have been encountered (62). Other patients have experienced an acute traumatic dislocation. In view of this catastrophic complication, children with Down syndrome should undergo a complete neurologic examination, including neuroimaging with lateral views of the cervical spine in flexion and extension, if they plan to participate in competitive sports (e.g., Special Olympics) or in sports that could result in trauma to the head or neck. Children who have this abnormality should be excluded from these activities. Children with atlantoaxial instability who develop neurologic signs require surgical stabilization of the joint (62,63).
In addition to these neurologic abnormalities, patients with Down syndrome exhibit a number of dysmorphic features. None of these is present invariably, nor are any consistently absent in the healthy population, but their conjunction contributes to the characteristic appearance of the patient.
The eyes show numerous abnormalities, almost all of which have been encountered in other chromosomal anomalies (64). The palpebral fissures are oblique (upslanting) and narrow laterally. Patients have a persistence of a complete median epicanthal fold, a fetal characteristic rarely present in healthy children older than age 10 years but observed in 47% of trisomy 21 patients aged 5 to 10 years and in 30% of older patients (65). Brushfield spots are an accumulation of fibrous tissue in the superficial layer of the iris. They appear as slightly elevated light spots encircling the periphery of the iris (Fig. 4.2). According to Donaldson, Brushfield spots are present in 85% of Down syndrome individuals, as compared with 24% of controls (66). Blepharitis and conjunctivitis are common, as are lenticular opacifications and keratoconus, particularly in older patients. Many children with Down syndrome benefit from spectacles to correct refractive errors and astigmatism.
Anomalies of the external ear are also frequent. The ear is small, low set, and often C-shaped, with a simple helix
and hypoplastic tragus. The cartilage is often deficient. Additionally, the diameter of the external auditory meatus is abnormally narrow, which precludes good visualization of the tympanic membrane.
and hypoplastic tragus. The cartilage is often deficient. Additionally, the diameter of the external auditory meatus is abnormally narrow, which precludes good visualization of the tympanic membrane.
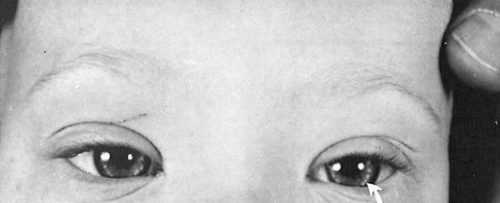 FIGURE 4.2. Five-year-old child with Down syndrome demonstrating Brushfield spots (arrow). (Courtesy of the late Dr. H. Zellweger, Department of Pediatrics, University of Iowa, Iowa City, IA.) |
Structural anomalies of the middle and inner ear might contribute significantly to the language delay commonly encountered in children with Down syndrome. Congenital malformations of the bones of the middle ear, permanent fixation of the stapes, and shortening of the cochlear spiral are encountered frequently (67). Additionally, children, particularly infants, are prone to acute and chronic otitis, middle ear effusions, and endolymphatic hydrops with subsequent conductive and neurosensory hearing loss.
According to Balkany and colleagues (68), 64% of patients with Down syndrome have binaural hearing loss. In 83%, the hearing loss is conductive, and in the remainder, sensorineural. Brainstem auditory-evoked potentials can be used to determine the type and severity of hearing loss in young or uncooperative children (69). Because of the frequency of impaired hearing, a comprehensive hearing evaluation is indicated during the first 6 to 12 months of life.
The lips of the patient with Down syndrome have radial furrows, and, as a consequence of the generalized hypotonia, the tongue, often fissured, tends to protrude because the maxilla is too small and the palate is too narrow to accommodate it. A typical finding, particularly evident in infants, is a roll of fat and redundant skin in the nape of the neck, which, combined with the short neck and low hairline, gives the impression of webbing.
The extremities are short. The fifth digit is incurved, and the middle phalanx is hypoplastic (clinodactyly). As a consequence, its distal interphalangeal crease is usually proximal to the proximal interphalangeal crease of the ring finger. Additionally, the distal and proximal interphalangeal creases are closely spaced, and there might be a single flexion crease on the fifth digits. The simian line, a single transverse palmar crease, is present bilaterally in 45% of patients (Fig. 4.3). In the feet, a diastasis between the first and second toes (sandal gap) is the most characteristic anatomic abnormality.
A juvenile rheumatoid arthritis–like progressive polyarthritis has been seen with some frequency. A number of other autoimmune disorders, notably diabetes mellitus type 1, also have a greater-than-chance association with Down syndrome, as do a variety of endocrine abnormalities (70). Down syndrome is associated with several autoimmune conditions and with a variety of endocrine abnormalities (71). An elevation of antithyroid antibody is common, and nearly 50% of trisomy 21 patients, mostly male patients, have reduced thyroid function in later life (72). Congenital hypothyroidism and primary gonadal deficiency are fairly common, but hypothyroidism can appear at any age. Gonadal deficiency progresses from birth to adolescence and becomes manifest in adult life with resultant infertility in virtually all nonmosaic male patients (73).
The dermatoglyphic pattern of the patient with Down syndrome shows several abnormalities. Of these, a distally displaced triradius, the palmar meeting point of three differently aligned fine creases, is seen in 88% of patients but in only 10% of controls (50). Dermatoglyphic abnormalities of the fingerprint patterns are equally characteristic (see Fig. 4.3) (50,74).
Abnormalities of the skin are common. Xeroderma and localized chronic hyperkeratotic lichenification are seen in 90% and 75% of patients, respectively. Vitamin A levels are reduced in 15% to 20% of patients.
As of 1997, the median age at death of Down patients was 49 years (75). Several factors contribute to the reduced life expectancy. The most important of these is the high incidence of major cardiovascular malformations; these account for 80% of deaths. In one study, 92.2% of Down syndrome patients without congenital heart disease survived to age 24 years, as contrasted with 74.6% of patients with congenital heart disease (76). As expected, mortality
in those with congenital heart disease is the greatest during the first year of life (77). Of less significance in terms of influencing life expectancy is a 10- to 20-fold increase in lymphoblastic leukemia (78). As a rule, acute myeloid leukemia predominates in children with Down syndrome younger than age 4 years, whereas acute lymphoblastic leukemia predominates in older children (78). Also shortening the life expectancy of children with Down syndrome is an unexplained high susceptibility to infectious illnesses and the early appearance of Alzheimer disease, which results in a marked decrease in survival after 35 years of age (75,79). The clinical picture of Alzheimer disease in patients with Down syndrome corresponds to that in patients without Down syndrome. Deterioration of speech and gait are early findings. Epileptic seizures and myoclonus are common and were seen in 78% and 85% of patients, repsectively, in the Dutch series of Evenhuis (80). Loss of cognitive function, including memory loss, generally remains unrecognized until the later stages of the disease.
in those with congenital heart disease is the greatest during the first year of life (77). Of less significance in terms of influencing life expectancy is a 10- to 20-fold increase in lymphoblastic leukemia (78). As a rule, acute myeloid leukemia predominates in children with Down syndrome younger than age 4 years, whereas acute lymphoblastic leukemia predominates in older children (78). Also shortening the life expectancy of children with Down syndrome is an unexplained high susceptibility to infectious illnesses and the early appearance of Alzheimer disease, which results in a marked decrease in survival after 35 years of age (75,79). The clinical picture of Alzheimer disease in patients with Down syndrome corresponds to that in patients without Down syndrome. Deterioration of speech and gait are early findings. Epileptic seizures and myoclonus are common and were seen in 78% and 85% of patients, repsectively, in the Dutch series of Evenhuis (80). Loss of cognitive function, including memory loss, generally remains unrecognized until the later stages of the disease.
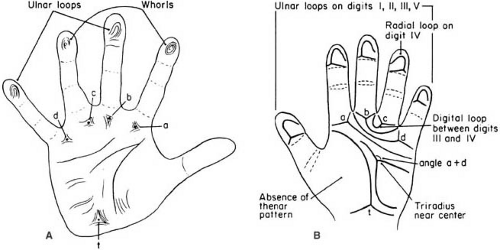 FIGURE 4.3. Outlines of the digital, palmar, and plantar configurations. A: Healthy child. The triradii are a, b, c, d, and t—the meeting point of sets of parallel ridges; t is located near the base of the fourth metacarpal bone at some point on its axis. B: Child with Down syndrome. A transverse palmar crease occurs, fingers tend to have a loop pattern, and the triradius subtends an angle of approximately 80 degrees with points a and d instead of the normal 45 degrees. (Modified from Penrose LS. Fingerprints, palms and chromosomes. Nature 1963;197:933.) |
Diagnosis
The diagnosis of Down syndrome is made by the presence of the characteristic physical anomalies and is confirmed by cytogenetic analysis. Karyotyping of all cases of Down syndrome is indicated to identify those that result from chromosomal translocation. When translocation is present, the karyotype of both parents must be ascertained to determine whether one of them carries a balanced translocation.
First-trimester screening is indicated for mothers ages 35 or older at term as well as in the young mother who has had a previous child with Down syndrome. Low maternal serum α-fetoprotein, reduction of unconjugated estriol, and elevated chorionic gonadotropins are independent predictors of Down syndrome, as is an ultrasonic measurement of the fetal nuchal translucency (81). Serum inhibin, a glycoprotein of placental origin, is elevated in maternal serum in women with Down syndrome fetuses and has been suggested as another useful screening marker (82), as is pregnancy-associated protein A (83). Biggio and coworkers discussed the cost-effectiveness, reliability, and complications of the various current screening strategies (84).
The diagnosis of a mosaic Down syndrome does not present any difficulty except when the frequency of the abnormal karyotype is low (85). However, accurate prediction of the phenotype in mosaic cases is problematic. Eventually, routine screening of all mothers for Down syndrome will be possible. One approach is the implementation of a polymerase chain reaction (PCR)–based technique that uses chromosome 21 markers to detect abnormal gene copy numbers in fetal cells (86,87).
Treatment
Despite the number of vigorously and fervently advocated regimens, none has proved to be successful in improving the mental deficit of the child with Down syndrome. Medical intervention is directed toward treatment and prophylaxis of infections, treatment of hearing and visual
deficits, early recognition and treatment of hypothyroidism, and, if possible, correction of congenital heart disease or any other significant malformation. Although not proven statistically, early intervention appears to be of benefit in enhancing communication and self-help skills.
deficits, early recognition and treatment of hypothyroidism, and, if possible, correction of congenital heart disease or any other significant malformation. Although not proven statistically, early intervention appears to be of benefit in enhancing communication and self-help skills.
The early administration of 5-hydroxytryptophan, a serotonin precursor, appears to reverse the muscular hypotonia characteristically present in the infant with Down syndrome (88), and in some instances can accelerate motor milestones. Unfortunately, this has no effect on the ultimate level of intelligence (89).
The treatment of the Down syndrome child with mental retardation is similar to that of children with mental retardation from any cause and having the same potential of intellectual and social function. The topic, particularly the role of early intervention and total communication programs, is discussed in Chapter 18.
Other Abnormalities of Autosomes
Autosomal trisomies, and even double autosomal trisomies, are seen in some 20% of spontaneously aborted fetuses, and only a small minority of trisomic conceptuses are live born. Aside from trisomy 21, trisomies involving the other, larger chromosomes are associated with multiple severe malformations of the brain and viscera, and affected children usually do not survive infancy. The two entities most commonly encountered by the pediatric neurologist are trisomies of chromosomes 13 and 18. The incidence of these and other chromosomal abnormalities is presented in Table 4.3 (90).
Trisomy 1 (Patau Syndrome)
Trisomy 13 (Patau syndrome) occurs in fewer than 1 in 5,000 births (91). Approximately one-half of cases are diagnosed prenatally through a combination of routine ultrasound and serum screening (92). The clinical picture is characterized by feeding difficulties, apneic spells during early infancy, and striking developmental retardation (93,94,95). Patients have varying degrees of median facial anomalies, including cleft lip and/or palate, premaxillary agenesis, and micrognathia. The ears are low set and malformed, and many infants are deaf. Polydactyly, flexion deformities of the fingers, and a horizontal palmar crease are common. The dermatoglyphics are abnormal (Fig. 4.4). Cardiovascular anomalies are present in 80% of patients, most commonly in the form of a patent ductus arteriosus or a ventricular septal defect. Approximately 33% of patients have polycystic kidneys. An increased frequency of omphalocele and neural tube defects also exists. Microcephaly is present in 83% and microphthalmus in 74% of cases (94).
The major neuropathologic abnormality is holoprosencephaly (arrhinencephaly), a complex group of malformations that have in common absence of the olfactory bulbs
and tracts and other cerebral anomalies, notably only one ventricle and absence of the interhemispheric fissure (see Chapter 5). On autopsy, a number of microscopic malformations are observed, including heterotopic nerve cells in the cerebellum and in the subcortical white matter (96,97,98).
and tracts and other cerebral anomalies, notably only one ventricle and absence of the interhemispheric fissure (see Chapter 5). On autopsy, a number of microscopic malformations are observed, including heterotopic nerve cells in the cerebellum and in the subcortical white matter (96,97,98).
TABLE 4.3 Incidence of Chromosomal Anomalies Among 3,658 Newborns in a Danish Population Between 1980 and 1982 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
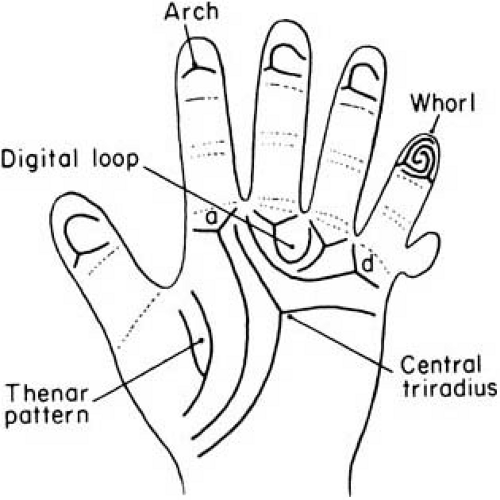 FIGURE 4.4. Dermatoglyphics in trisomy 13. Polydactyly is present. The angle subtended by points a, d, and the central triradius is almost 90 degrees. Unlike in Down syndrome, both arches and whorls can be present. (Modified from Penrose LS. Fingerprints, palms and chromosomes. Nature 1963;197:933.) |
Absence of the olfactory apparatus is not specific for trisomy 13. In the series of Gullotta and coworkers (99), this malformation was encountered at autopsy in 40% of trisomy 13 patients. It also occurs with deletion of the short arm of chromosome 18 (18p), in the trisomy 18 syndrome, and with deletion of the long arm of chromosome 13 (13q). A condition termed pseudotrisomy 13 syndrome is fairly common. This entity is characterized by alobar holoprosencephaly, cardiac anomalies, and postaxial polydactyly, accompanied by apparently normal cytogenetics (100). Cardiac, pulmonary, genital, or other skeletal malformations also can be present (101). The condition is transmitted as an autosomal recessive trait (102).
The prognosis for children with chromosome 13 trisomy is poor. Approximately 50% die during the first month of life, and only 20% survive into the second year.
Trisomy 2 (Edwards Syndrome)
Trisomy 18 (Edwards syndrome) was first described in 1960 by Edwards (103). Its incidence appears to be on the increase; with the most recently recorded prevalence in Hawaii being 4.7 in 10,000 (91). The incidence in conceptuses is much higher, and only 2.5% of trisomy 18 conceptuses are live born. The anomaly is seen much more frequently in girls, the most recent male:female ratio being 0.65 (104). This suggests that a high proportion of affected male fetuses succumb during gestation. Like other autosomal trisomies, trisomy 18 results from an error in maternal meiosis, with the additional chromosome demonstrating maternal origin in 96% of cases (105).
The clinical picture is highlighted by a long, narrow skull with prominent occiput, characteristic facies, low-set, malformed ears, a mildly webbed neck, and marked physical and mental retardation (94,99). A typical picture of camptodactyly (flexion contractures of the fingers) and abnormal palmar flexion creases occurs. The second and fourth fingers characteristically overlie the third, and the fifth finger overlies the fourth. Distally implanted, retroflexible thumbs are encountered frequently. As in trisomy 13, omphalocele, polycystic kidneys, and congenital heart disease are common. A striking dermatoglyphic pattern, relatively less common in other autosomal anomalies, is the presence of simple arches on all fingers.
No consistent central nervous system malformation is associated with the trisomy 18 syndrome. The most common anomaly is agenesis of the corpus callosum, occasionally associated with nerve cell heterotopias of the cerebellum or white matter (97,99). In more than 50% of patients, the brain is small, but normal on gross and routine microscopic examination (99). The prognosis for children with trisomy 18 is poor, and 90% die by 1 year of age. The mean survival time is 10 months for girls and 23 months for boys (106). The natural history of trisomy 18 and trisomy 13 in long-term survivors has been well characterized (106,107).
Other trisomies are much less common (see Table 4.3) but have been found with relatively high frequency in spontaneous human abortions (9). Mosaic trisomy syndromes have been recognized for chromosomes 8, 9, 16, and 20. Mosaicisms for other autosomal trisomies have been reported very rarely (108). Their clinical appearance is reviewed by Schinzel (12) and Tolmie (109).
STRUCTURAL AUTOSOMAL ANOMALIES
The best described of the numerous classical deletion syndromes are the 5p- (cri du chat), and the 4p- (Wolf-Hirschhorn) syndromes.
Cri du Chat Syndrome
A number of cytogenetically visible structural anomalies of the autosomal chromosomes have been reported and have been accompanied by neurologic defects, notably mental retardation and microcephaly. The most common of these entities is a deletion of the short arm of chromosome 5 (5p), which causes the cri du chat syndrome (110). Through the combined phenotypic and molecular analysis of affected individuals the genes responsible for this condition have been mapped to 5p15.2 and 5p15.3. Deletion of a critical region in 5p15.2 results in the characteristic facial features and the severe mental retardation, whereas deletion of 5p15.3 is associated only with the meowing cry (111). The telomerase reverse transcriptase gene is localized to the region that is lost in cri du chat syndrome. The phenotypic effects of haploinsufficiency for this gene are unknown (112).
Cri du chat syndrome is seen with a frequency of 1 in 20,000 to 50,000 births (22,110). A low birth weight and failure in physical growth are associated with a characteristic cry likened to that of a meowing kitten. Infants have microcephaly, large corneas, moon-shaped facies, ocular hypertelorism, epicanthal folds, transverse palmar (simian) creases, and generalized muscular hypotonia. The brain, although small, is grossly and microscopically normal, representing a rare example of true microcephaly (113).
Wolf-Hirschhorn Syndrome
The Wolf-Hirschhorn syndrome is caused by partial monosomy of the short arm of chromosome 4, with the region
critical for the development of this disorder being 4p16.3 (114,115). The syndrome is characterized by a facies that has been likened to that of a helmet of a Greek warrior. Frontal bossing, a high frontal hairline, a broad, beaked nose, prominent glabella, and ocular hypertelorism are seen. Major malformations occur commonly, especially cleft lip and palate, and mental retardation is usually severe. Deletions in the region responsible for Wolf-Hirschhorn syndrome overlap those seen in Pitt-Rogers-Danks syndrome. The latter entity has a somewhat different phenotype. It is marked by prenatal and postnatal growth retardation, microcephaly, prominent eyes, a large mouth, and maxillary hypoplasia. Mental retardation is somewhat less severe than is seen in the Wolf-Hirschhorn syndrome. Wright and coworkers proposed that the two conditions result from the absence of similar, if not identical genetic segments, and that the clinical differences between the two syndromes result from allelic variation in the remaining homologue (116).
critical for the development of this disorder being 4p16.3 (114,115). The syndrome is characterized by a facies that has been likened to that of a helmet of a Greek warrior. Frontal bossing, a high frontal hairline, a broad, beaked nose, prominent glabella, and ocular hypertelorism are seen. Major malformations occur commonly, especially cleft lip and palate, and mental retardation is usually severe. Deletions in the region responsible for Wolf-Hirschhorn syndrome overlap those seen in Pitt-Rogers-Danks syndrome. The latter entity has a somewhat different phenotype. It is marked by prenatal and postnatal growth retardation, microcephaly, prominent eyes, a large mouth, and maxillary hypoplasia. Mental retardation is somewhat less severe than is seen in the Wolf-Hirschhorn syndrome. Wright and coworkers proposed that the two conditions result from the absence of similar, if not identical genetic segments, and that the clinical differences between the two syndromes result from allelic variation in the remaining homologue (116).
TABLE 4.4 Some Structural Autosomal Anomalies Associated With Neurologic Symptoms | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
Other structural autosomal anomalies associated with neurologic symptoms are summarized in Table 4.4.
Partial deletions and partial trisomies have been described for almost every chromosome, but few of these entities are sufficiently characteristic to allow the practitioner to suspect the diagnosis without cytogenetic analysis. In particular, the phenotypic expressions of ring chromosomes overlap those of partial deletions, showing so much variability that one cannot arrive at any conclusions with respect to their clinical pictures. Furthermore, certain features, especially microcephaly, are common to many of the chromosomal anomalies, and they also are encountered in the absence of any discernible chromosomal defect. Therefore, only those anomalies that occur with sufficient frequency to establish a fairly definite clinical
picture are discussed here. The most complete review of the various neurologic deficits seen in chromosomal disorders is that by Kunze (134).
picture are discussed here. The most complete review of the various neurologic deficits seen in chromosomal disorders is that by Kunze (134).
TABLE 4.5 Subtelomeric Deletions | ||
|---|---|---|
|
SUBTELOMERIC ABNORMALITIES
Deletions, duplications, and cryptic imbalance rearrangements of the telomeres—the terminal segments of chromosomes—have received an increasing amount of attention in the last few years. It has become apparent that abnormalities in these regions are responsible for at least 5% to 10% of nonsyndromic mental retardation as well as for cases of mental retardation associated with multiple congenital anomalies. Several techniques have been used to screen for these abnormalities. Fluorescence in situ hybridization (FISH) is being replaced by comparative genomic hybridization (CGH). In this method an abnormal and a control genome are compared for DNA copy differences. This technique is being adapted for rapid and sensitive automation (135,136,137,138). The recognized subtelomeric deletions are listed in Table 4.5.
SEX CHROMOSOME ABNORMALITIES
Sex chromosome abnormalities consist of various aneuploidies involving the sex chromosomes. As determined by most surveys of the newborn population (139a) or of cells obtained at amniocentesis (140), the incidence of sex chromosome abnormalities is approximately 2.5 in 1,000 phenotypic male infants and 1.4 in 1,000 phenotypic female infants (139a). Although considerable geographic variation occurs, the two most common abnormalities in phenotypic male patients are the XYY and the XXY (Klinefelter) syndromes (139a). Because monosomy X (45,X) is highly lethal, a significantly lower incidence of sex chromosome abnormalities is found in newborns than is found at amniocentesis, and in a large proportion of individuals with sex chromosome abnormalities, particularly phenotypic male individuals, the condition remains undiagnosed throughout life (140,141). The great majority of the abnormalities in female individuals (75%) involve the XXX syndrome, with less than 25% being monosomy X (Turner syndrome) or variants thereof (142).
Klinefelter Syndrome
Klinefelter syndrome was first described by Klinefelter in 1942 in male patients who suffered from increased breast development, lack of spermatogenesis, and increased excretion of follicle-stimulating hormone. The chromosome constitution of XXY, present in most patients, was elucidated in 1959 (143).
The condition occurs in 1 in 500 to 1,000 male births (142). Although affected persons are relatively tall with an eunichoid habitus, the diagnosis is usually not made until adult life, and hormone assays are normal before the onset of puberty (144). Neurologic examination shows decreased muscle tone and some dysmetria or tremor; the major deficits encountered in unselected patients are cognitive disorders. In the Denver series, IQs were 10 points lower than in controls (145), whereas in the Edinburgh series of Ratcliffe, the mean IQ of XXY patients was 96, as contrasted with a mean IQ of 105 for controls (146). A high incidence of impaired auditory sequential memory occurs, which results in delayed language development and a variety of learning disorders, notably dyslexia (147). An increase in the incidence of behavioral abnormalities also has been observed (146). EEG abnormalities, including spike and spike-wave discharges, are said to be common (148). On high-resolution magnetic resonance imaging (MRI) left temporal lobe gray matter is significantly reduced and brain volume is less than that of controls (149,150). A greater-than-chance association exists between Klinefelter syndrome and midline suprasellar or pineal germinomas (151), male breast cancer, and insulin-resistant diabetes.
XYY Karyotype
The XYY karyotype is another common chromosomal anomaly encountered in the phenotypic male individual, with a frequency as high as 4 in 1,000 male newborns (152).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


