Chapter 31 Chromosomes and Chromosomal Abnormalities
This chapter focuses on the approach to chromosomal disorders in pediatric neurology. The various methods of chromosomal analyses are considered first, followed by a description of the various types of chromosomal abnormalities. This discussion is followed by an overview of the clinical approach to chromosomal abnormalities, and then a brief clinical description of chromosomal syndromes relevant to the practice of pediatric neurology. The chapter closes with a look at the future of cytogenetic analysis (see also chapter 30).
Methods of Chromosome Analysis
Chromosome Banding
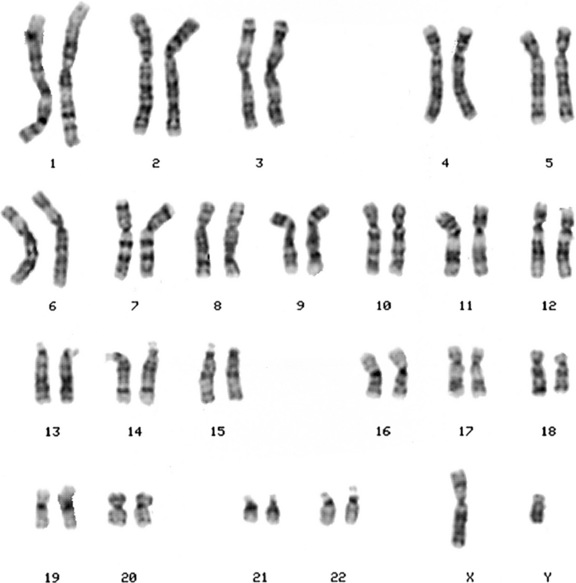
Chromosomes are displayed as a karyotype (Figure 31-1), which is prepared by arranging homologous chromosomes in an orderly fashion, starting from chromosome 1 and ending with chromosome 22, as well as the sex chromosomes. Subsequent developments in laboratory cytogenetics have gradually improved the resolution of chromosomal analysis. As the cell proceeds through mitosis, the chromosome gradually contracts, until anaphase, when the chromatids separate. If cells are collected during early prophase, chromosomes are highly extended, revealing a fine, highly detailed banding pattern. This banding pattern has facilitated recognition of subtle chromosome rearrangements involving small chromosome segments. Even with this approach, however, the resolution is limited to 3–5 million bp (Mb) of DNA, which may include dozens of genes.
Molecular Cytogenetics
The gap between light microscope resolution of chromosome structure and the gene was bridged by the introduction of several molecular cytogenetic techniques. Fluorescence in situ hybridization (FISH) involves hybridizing a fluorescently labeled single-stranded DNA probe to denatured chromosomal DNA on a microscope slide preparation of metaphase chromosomes and/or interphase nuclei prepared from the patient’s sample. After overnight hybridization, the slide is washed and counterstained with a nucleic acid dye (e.g., DAPI, or 4′,6-diamidino-2-phenylindole), allowing the region where hybridization has occurred to be visualized using a fluorescence microscope. FISH is now widely used for clinical diagnostic purposes. There are different types of FISH probes, including locus-specific probes, centromeric probes, and whole-chromosome paint probes. Locus-specific probes are specific for a particular single locus. They are especially useful for identifying subtle submicroscopic deletions and duplications (Figure 31-2). Centromeric probes are specific for unique repetitive DNA sequences (e.g., alpha-satellite sequences) in the centromere of a specific chromosome. They are suitable for making a rapid diagnosis of one of the common aneuploidy syndromes (trisomies 13, 18, and 21, and sex chromosome aneuploidies) using nondividing interphase nuclei. This is particularly useful in a prenatal setting using amniotic fluid or chorionic villi samples (CVS). Whole-chromosome paint probes consist of a cocktail of probes obtained from different regions of a particular chromosome. When this cocktail mixture is used in a single hybridization, the entire relevant chromosome fluoresces (is “painted”). Whole-chromosome paints are useful for characterizing complex chromosomal rearrangements, and for identifying the origin of additional chromosomal material, such as small marker or ring chromosomes.
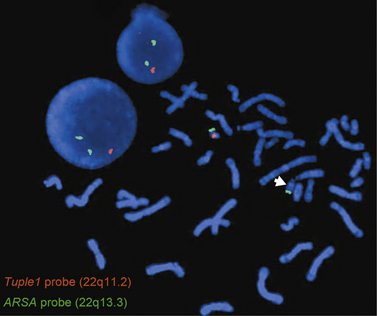
FISH using locus-specific probes has been extremely useful in the detection of “microdeletion syndromes” resulting from deletions of multiple contiguous genes. These are subtle submicroscopic deletions that are below the resolution of the routine G-banded chromosome analysis. Also, two-color and three-color FISH applications are routinely used to diagnose specific deletions, duplications, or other rearrangements, both in metaphase chromosomes and in interphase nuclei. Use of FISH usually requires that the patient either exhibits features consistent with a well-defined syndrome with known chromosomal etiology, or demonstrates an abnormal karyotype. This is because single FISH probes reveal rearrangements only of the segments being interrogated, but do not provide information about the rest of the genome. Another limitation of FISH is the number of probes that can be applied in a simultaneous assay. FISH techniques have been developed utilizing pools of whole-chromosome paint probes for every chromosome to provide a multicolor human karyotype in which each pair of homologous chromosomes can be identified on the basis of its unique color when studied using special computer-based image analysis software (spectral karyotyping and multicolor or M-FISH) [Liehr et al., 2004].
One type of FISH that has the potential to reveal chromosomal imbalances across the genome is comparative genomic hybridization (CGH). In CGH, DNA specimens from the patient and a normal control are differentially labeled with two different fluorescent dyes and hybridized to normal metaphase chromosome spreads. Difference between the fluorescent intensities of the two dyes along the length of any given chromosome will reveal gains and losses of genomic segments [Levy et al., 1998]. The limitations of this technology include many of the same limitations of G-banded chromosome analysis. Thus, like G bands, the resolution of CGH is limited to that of metaphase chromosomes, which is approximately 5 Mb for most clinical applications [Liehr et al., 2004].
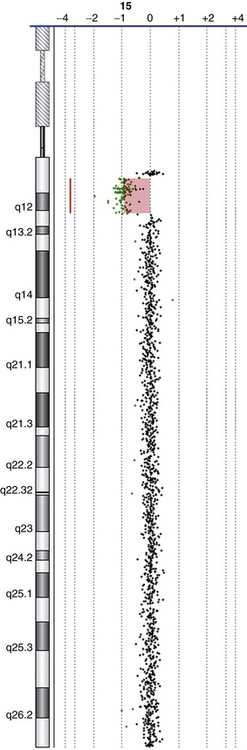
The latest addition to molecular cytogenetic techniques is array CGH, where CGH is applied to an array of DNA targets (probes), each representing a part of the human genome and fixed to a solid support (usually a glass slide). Like CGH, array CGH directly compares DNA content between two differentially labeled DNA specimens (a test or patient, and a reference or normal control), which are labeled and co-hybridized on to the array. Arrays have been constructed with a variety of DNA targets, ranging from bacterial artificial chromosomes (BACs), which are 80–250 thousand bp (kb) long [Shaffer and Bejjani, 2006] to oligonucleotides (oligos), which are 25–80 bp long [Lucito et al., 2003; Ylstra et al., 2006]. Following hybridization and washing to remove unbound DNA, the array is scanned and analyzed using special computer software to measure the relative ratios of fluorescence of the two dyes, and detect gains/losses of genomic regions represented on the array (Figure 31-3). The resolution of array CGH is dependent on the type of probes used (BACs or oligos) and the distance between them. In the past few years, high-resolution whole-genome coverage array CGH platforms have been increasingly used in clinical molecular cytogenetic labs. These provide a relatively quick method of scanning the entire genome for gains and/or losses with significantly higher resolution and greater clinical abnormality yield than was previously possible. This led to the identification of novel genomic disorders in patients with autistic spectrum disorders (ASD), developmental delay (DD), mental retardation (MR), and/or multiple congenital anomalies (MCAs) [Edelmann and Hirschhorn, 2009].
Chromosomal Abnormalities
Deletions and Duplications
A deletion involves loss of part of a chromosome and results in monosomy for that segment of the chromosome, whereas duplication represents the doubling of part of a chromosome, resulting in trisomy for that segment. The result is either decrease (in a deletion) or increase (in a duplication) in gene dosage. In general, duplications appear to be less harmful than deletions. Very large deletions usually are incompatible with survival to term. Deletions or duplications larger than approximately 5 Mb in size can be visualized under the microscope using G-banded chromosome analysis. Clinical syndromes resulting from submicroscopic deletions or duplications (i.e., microdeletions/microduplications) with a size <5 Mb have been identified with the help of molecular cytogenetic techniques, including FISH and array CGH. In these syndromes, groups of contiguous genes are either deleted or duplicated, resulting in a defined set of congenital anomalies. The molecular mechanisms responsible for these microdeletions/microduplications have been extensively studied and are well documented [Gu et al., 2008]. Specific microdeletion/microduplication syndromes of neurologic interest are described later in this chapter.
Translocations
Translocations involve the exchange of genetic material between chromosomes. In a balanced reciprocal translocation the exchange is equal, with no loss or gain of genetic material, though it is possible for a gene to be disrupted at one of the breakpoints. More often, the carrier of a balanced translocation is free of clinical signs or symptoms but is at risk for having offspring with unbalanced chromosomes. The risk for production of unbalanced gametes from a balanced translocation carrier depends on the chromosomes involved, the specific breakpoints of the translocation, and the sex of the carrier. Empirical data are available for some specific translocations [Daniel et al., 1989]. Risks include miscarriage and birth of a liveborn child with congenital anomalies, resulting from chromosome imbalance. The phenotype usually is a complex mixture of the results of loss or gain of at least two chromosome segments and therefore can be difficult to predict.
Marker and Ring Chromosomes
A “marker” chromosome is a rearranged chromosome whose genetic origin is unknown based on its G-banded chromosome morphology. Usually, these chromosomes are present in addition to the normal chromosome complement and are thus called supernumerary marker chromosomes (SMCs). The birth prevalence of SMCs is in the range of 2–7 per 10,000, and 30–50 percent originate from chromosome 15 [Gardner and Sutherland, 2004]. Two-thirds of de novo marker chromosomes can be associated with an abnormal outcome, whereas inherited ones can be passed from generation to generation without apparent clinical effects. Larger markers with more genetically active material are more likely to be of clinical significance. FISH and array CGH have proved very helpful in the precise identification of the genetic origin of SMCs.
Isochromosomes
An “isochromosome” is a chromosome in which one arm is missing and the other duplicated in a mirror-image fashion. The most probable mechanism for the formation of an isochromosome is the misdivision through the centromere in meiosis II, wherein the centromere divides transversely rather than longitudinally. The most commonly encountered isochromosome is that which consists of two long arms of the X chromosome. This accounts for approximately 15 percent of all cases of Turner’s syndrome [Gardner and Sutherland, 2004].
Cytogenetic Nomenclature
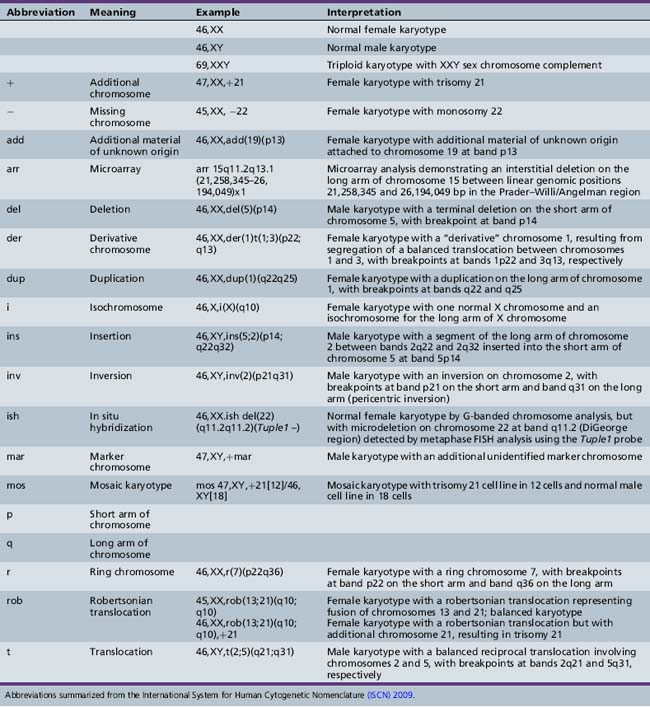
By convention, each chromosome arm is divided into regions, and each region is subdivided into bands and sub-bands, numbered from the centromere outwards. Cytogeneticists describe findings of chromosomal analysis using a standardized system of nomenclature (International System for Human Cytogenetic Nomenclature). Detailed description of this system is beyond the scope of this chapter, but major terms with examples are listed in Table 31-1. The normal male karyotype is designated 46,XY and the normal female karyotype is 46,XX. Any chromosomal abnormality is described after the sex chromosome constitution.
Incidence of Chromosomal Abnormalities
Estimates of the incidence of chromosomal abnormalities vary with the mode of ascertainment and the technology used for chromosome analysis. In general, the incidence falls rapidly from conception to birth. The highest rates have been observed among products of conception from first-trimester spontaneous abortions. Approximately 50 percent of these spontaneous miscarriages have a chromosomal abnormality [Boue et al., 1973]. By birth, the rate of chromosomal abnormalities declines to approximately 0.5–1 percent in liveborn infants, although the rate is much higher (5–10 percent) in stillborn infants [Jacobs et al., 1992].
Clinical Indications for Cytogenetic Analysis
Multiple Congenital Anomalies
Genetic imbalance resulting from a chromosomal abnormality usually leads to aberrant embryonic development. Most commonly, this abnormal development involves multiple tissues, including the brain. Many specific syndromes can be recognized from a constellation of dysmorphic physical features and specific congenital anomalies. The clinician should be familiar with the most common syndromes, especially those resulting from trisomies 13, 18, and 21, as well as the sex chromosome aneuploidies (47,XXX, 47,XXY, and 45,X). Phenotypes resulting from duplication or deletion of smaller amounts of genetic material can be more difficult to identify clinically. Some of the more important syndromes are described in the next section. Some clues to the occurrence of a chromosomal abnormality are provided in Box 31-1. As a rule, chromosomal studies should be performed in a patient who exhibits congenital anomalies involving two or more tissues, in whom a specific alternative diagnosis cannot be established, and if the anomalies are not related to one another as cause and effect (e.g., hydrocephalus resulting from spina bifida).










Developmental Delay and/or Mental Retardation
In some chromosomal abnormalities, the phenotype is primarily that of developmental delay (DD) and/or mental retardation (MR), with few or no congenital anomalies. Sometimes, minor dysmorphic features are present, but these often are not noticed on routine examination. G-banded chromosome analysis and array CGH testing therefore should be considered in the evaluation of a child with unexplained DD/MR. MR is a common condition that affects 1–3 percent of the population, and the cause is established in only 50 percent of the cases [Anderlid et al., 2002; Kriek et al., 2004] (also see chapter 43).
The use of array CGH to analyze the genomes of normal humans led to the discovery of extensive genomic copy number variations (CNVs), both gains and losses, ranging in size from kb to Mb, and not recognized by high-resolution G-banded chromosome analysis [Iafrate et al., 2004; Sebat et al., 2004]. CNVs have been proposed to be a major factor responsible for human diversity [Lupski, 2006]. Through genomic rearrangement of rearrangement-prone regions as a result of the genomic architecture, CNVs can cause genomic disorders due to gains and/or losses of dosage-sensitive gene(s), resulting in a clinical phenotype [Stankiewicz and Beaudet, 2007]. Using array CGH technologies, clinically significant pathogenic CNVs have been reported in up to 17 percent of patients with ASD, DD, MR, and/or MCAs [Stankiewicz and Beaudet, 2007; Edelmann and Hirschhorn, 2009].
Fertility Problems
Chromosomal imbalance most often leads to miscarriage rather than to live birth. Carriers of balanced rearrangements, including translocations or inversions, may therefore come to attention through recurrent miscarriage [Flint and Gibb, 1996; Hook and Cross, 1989]. It is recommended that couples who have experienced two or more unexplained first-trimester miscarriages be offered chromosomal analysis. Finding a balanced rearrangement permits genetic counseling of the couple, including offering prenatal diagnosis for future pregnancies. Other members of the family also may carry the balanced rearrangement and should be offered counseling and testing. Unexplained infertility should prompt a request for chromosome studies, especially for women presenting with primary amenorrhea, and for men presenting with azoospermia.
Prenatal diagnosis
Chromosomal analysis of a developing fetus can be achieved through collection of fetal cells by CVS, amniocentesis, or peripheral umbilical blood sampling (PUBS) [D’Alton and DeCherney, 1993]. CVS involves sampling part of the fetal placenta using a biopsy device either passed through the cervix or inserted by a needle through the mother’s abdomen [Pijpers et al., 1988; Smidt-Jensen and Hahnemann, 1988]. It is performed at 10–12 weeks of gestation. CVS offers the advantage of early testing. Amniocentesis involves sampling amniotic fluid at 16–18 weeks of gestation. Fetal cells in the fluid are cultured and can be used for chromosomal analysis. PUBS is offered after 20 weeks of gestation and involves sampling fetal blood by nicking the umbilical vein under ultrasound guidance [Sermon et al., 2004].
Indications for prenatal testing are listed in Box 31-2. General practice is to offer prenatal testing for pregnancies in which the risk of a chromosomal abnormality exceeds the risk of a complication of the procedure. For couples in which one partner carries a chromosome rearrangement, prenatal testing to detect unbalanced chromosomes can be offered. The actual risk of unbalanced chromosomes in the pregnancy depends on the nature of the rearrangement but generally is greater than 1 percent. The laboratory performing the prenatal testing must be informed of the details of the rearrangement, to ensure that subtle changes are detected. The recurrence risk for future trisomy for a couple who have had one pregnancy affected with trisomy is approximately 1 percent [Lister and Frota-Pessoa, 1980]. This risk is irrespective of the particular chromosome involved in the trisomy. Pregnancies are increasingly being monitored for fetal anomalies by ultrasound or maternal serum screening, with findings indicative of increased risk followed up by prenatal diagnostic testing.







Specific Cytogenetic Syndromes
Polyploidy
Clinical Features
The triploid phenotype is distinct and easily recognized. Polyhydramnios or pre-eclampsia may complicate the pregnancy. The placenta may be large, and hydatidiform changes may be seen. Birth weight usually is low. Syndactyly involving the third and fourth digits is characteristic. Craniofacial features include low-set, malformed ears, hypertelorism, and micrognathia. Cardiac, renal, and central nervous system malformations are common. Males may have dysplastic external genitalia. Studies of the parental origin of the three chromosome sets in triploidy have revealed that a majority of affected persons have two maternal sets, perhaps because of more frequent survival to term of triploid fetuses with two maternal sets of chromosomes (digynic triploids) [McFadden et al., 1993]. Long-term survivors often are mosaics and may have less obvious phenotypic features. Body asymmetry and pigmentary dysplasia may be clues to chromosomal mosaicism in general, including, in some cases, triploidy [Woods et al., 1994].
Trisomy 13 (Patau Syndrome)
Cytogenetics
Trisomy 13 occurs in approximately 1 in 7000 live births [Savva et al., 2010]. A majority of affected persons have 47 chromosomes, with an extra copy of chromosome 13. Approximately 5–10 percent have trisomy because of translocation between 13 and another acrocentric chromosome, usually chromosome 14 (robertsonian translocation). Mosaicism occurs in a small proportion of cases and may ameliorate the phenotype. Duplication of part of chromosome 13 resulting from unbalanced translocation can result in abnormal phenotypic features, although not necessarily similar to those seen in full trisomy 13. Advanced maternal age has been a factor in the occurrence of this aneuploidy syndrome.
Clinical Features
Trisomy 13 is associated with congenital anomalies involving most major organ systems (Figure 31-4). Holoprosencephaly is the hallmark central nervous system anomaly [Moerman et al., 1988], occurring in about 80 percent of cases. Infants with trisomy 13 who demonstrate holoprosencephaly usually have accompanying craniofacial anomalies. The eyes may be set closely together (hypotelorism) or even fused in a single orbit (cyclopia). Other ocular anomalies include micro-ophthalmia, iris colobomata, cataracts, and retinal dysplasia. Premaxillary agenesis and cleft lip or palate also may be present. Ulcer-like defects in scalp skin (cutis aplasia) occur commonly. Limb anomalies include postaxial polydactyly in two-thirds of patients and rocker-bottom foot. Congenital heart defects, especially ventricular septal defect (VSD), are common, as are renal anomalies, including cystic dysplasia. The phenotype overlaps to some degree with that of Meckel–Gruber syndrome (encephalocele, polydactyly, polycystic kidney), inherited as an autosomal-recessive trait due to mutation of the MKS1 gene. This overlap underlines the importance of confirming the clinical diagnosis of trisomy 13 by chromosomal analysis.
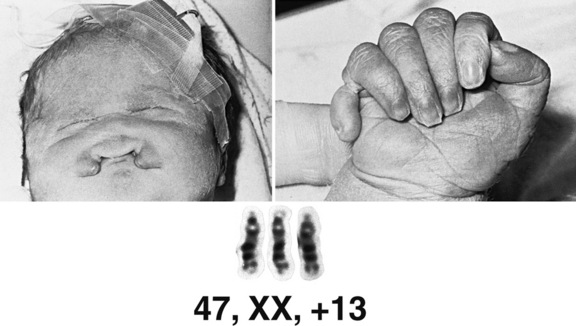
Management
Few infants with trisomy 13 survive the newborn period, with apnea being the most common cause of death [Rasmussen et al., 2003]. Often the anomalies are too numerous and severe to be corrected. In the absence of life-threatening malformations, however, long-term survival has been well documented, albeit usually with severely impaired cognitive function. Baty and co-workers documented the natural history of this disorder [Baty et al., 1994; Baty et al., 1994a & b].
Trisomy 18 (Edwards’ Syndrome)
Clinical Features
Infants with trisomy 18 have low birth weight and microcephaly. Other common features include a prominent occiput, low-set “simple” ears, and a small mouth (Figure 31-5). Hands usually are tightly clenched in a characteristic configuration, with the fourth and fifth fingers overlapping the first and second. Terminal phalanges often are hypoplastic, and rocker-bottom foot may be present. Congenital heart defects and renal anomalies also are common. Brain malformations include heterotopias, agenesis of the corpus callosum, Dandy–Walker malformation, and Arnold–Chiari malformation. Infants commonly are jittery and hypertonic, and have apnea and seizures.
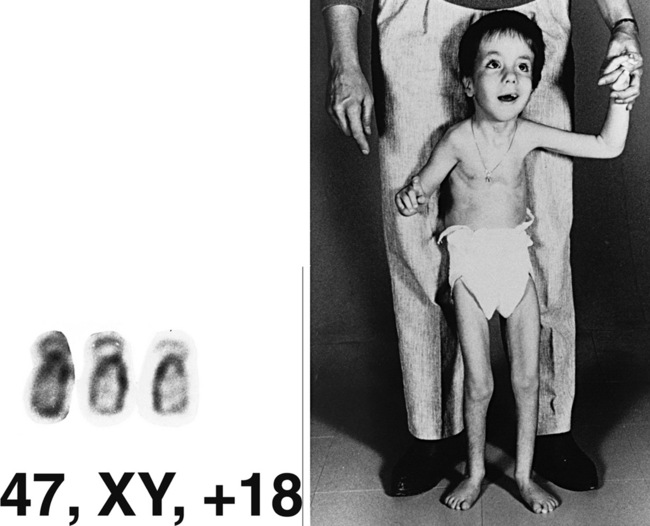
Fig. 31-5 A patient with trisomy 18 at 7 years of age.
(Karyotype courtesy of M Rochon, Sherbrooke, Quebec, Canada.)

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


