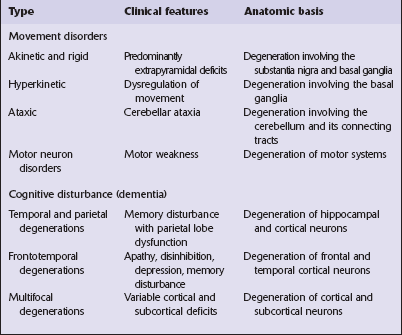
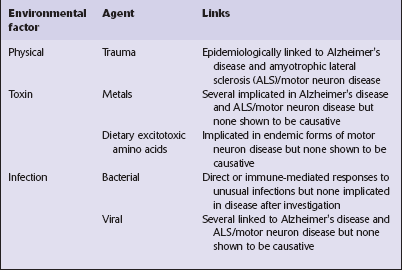
26 There are two main groups of neurodegenerative diseases: the movement disorders and the dementia syndromes (Table 26.1). Most neurodegenerative diseases are of unknown cause, but recent work has provided some insights into the mechanisms of neurodegeneration. Many environmental factors, such as toxins and viruses, have in the past been considered as possible causes of neurodegenerative diseases (Table 26.2), but the search for such factors has been largely disappointing and none has been established as causative. The prion diseases (see Chapter 32) represent a biological phenomenon that is probably unique in which a protein-only transmissible agent is responsible for disease.
Classification and pathogenesis of neurodegenerative diseases
CLASSIFICATION
 The clinical features of Parkinson’s disease are most commonly due to Lewy body pathology, but can also be caused by multiple system atrophy or by several disorders characterized by neurofibrillary tangle formation.
The clinical features of Parkinson’s disease are most commonly due to Lewy body pathology, but can also be caused by multiple system atrophy or by several disorders characterized by neurofibrillary tangle formation.
PATHOGENESIS
PROTEIN ACCUMULATION AND DEGRADATION
 an affinity for Congo red (and related stains)
an affinity for Congo red (and related stains)
 a yellow–green birefringence when the stained material is viewed under polarized light.
a yellow–green birefringence when the stained material is viewed under polarized light.![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Classification and pathogenesis of neurodegenerative diseases






Only gold members can continue reading. Log In or Register to continue


