CLCN2 and Idiopathic Generalized Epilepsy
Armin Heils
Clinic of Epileptology and Institute of Human Genetics, University of Bonn, Bonn, Germany
Introduction
Epilepsy is one of the most frequent neurologic diseases affecting about 3% of the worldwide population (1). Clinical symptoms range from mild drowsiness to severe generalized convulsions and are induced by synchronous neuronal discharges due to an imbalance of inhibitory and excitatory activity in the brain. Idiopathic forms are genetically determined and account for about 40% of all epilepsies (2,3). They are defined by recurrent seizures with characteristic clinical and electroencephalographic features in the absence of any detectable brain lesion. The most frequent idiopathic form of epilepsy is idiopathic generalized epilepsy (IGE), which comprises seven clinically delineated syndromes with age-related onset (4). The most common IGE subtypes are childhood and juvenile absence epilepsy (CAE, JAE), juvenile myoclonic epilepsy (JME), and epilepsy with grand mal seizures on awakening (EGMA) (4). Absence seizures are the leading symptom of CAE and JAE, characterized by a brief loss of consciousness (usually 10–20 s) and either manifest during childhood (CAE) or adolescence (JAE). JME manifests in adolescence with bilateral myoclonic jerks of arms and shoulders (myoclonic seizures) usually occurring in the early morning without a loss of consciousness. All types of IGE can be associated with generalized tonic–clonic seizure (GTCS), which typically occur on awakening, often provoked by sleep deprivation. When this is the only seizure type, patients are diagnosed with EGMA. In all subtypes of IGE, typical electroencephalographic (EEG) features are generalized spike-wave or polyspike-wave discharges, reflecting a state of synchronized neuronal hyperexcitability.
Several genes have already been identified to cause monogenic forms of idiopathic epilepsy. Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) has been shown to arise from mutations identified in genes encoding the α4 or β2 subunit of neuronal nicotinic acetylcholine receptors (CHRNA4, CHRNB2). Mutations in two voltage-gated potassium channel genes (KCNQ2, KCNQ3) are associated with benign familial neonatal convulsions (BFNC). Mutations in three different sodium channel subunits (SCN1B, SCN1A, SCN2A) as well as in the γ2 subunit of the GABAA receptor (GABRG2) have been shown to cause generalized epilepsy with febrile seizures plus (GEFS+) (see 5,6,7 for reviews). Thus, all genes identified in idiopathic epilepsies so far encode ion channels. Recently, autosomal dominant partial epilepsy with auditory features, an idiopathic form of temporal lobe epilepsy, has been shown to be caused by mutations in the leucine-rich, glioma-inactivated 1 gene (LGI1), not encoding an ion channel (8). Regarding the genetic basis of pure forms of the common IGE subtypes, thus far only one mutation in another GABAA receptor subunit gene (GABRA1) has been reported in a family with JME (9). A single epilepsy gene whose mutations can cause the whole spectrum of common IGE subtypes has not been identified to date.
Genome Scan for Common Idiopathic Generalized Epilepsy Loci
Recently, a genome wide search for chromosomal susceptibility loci of common subtypes including 130 IGE multiplex families identified a novel susceptibility locus on chromosome 3q26 (Z NPL = 4.19 at D3S3725; P = 0.000017) (10). One of the candidate genes located in this chromosomal region is CLCN2, which encodes the voltage-gated chloride channel ClC-2 (11,12). ClC-2 is strongly expressed in the brain, particularly in γ-aminobutyric acid (GABA)-inhibited neurons (13,14).
Several experimental results suggest an important role of this channel in establishing and maintaining a low intracellular chloride concentration ([Cl−]i), which is necessary for an inhibitory GABA response. When hippocampal pyramidal neurons—the best studied model for GABAergic synaptic inhibition in the brain (15,16,17) are loaded with chloride and when K-Cl cotransport is simultaneously blocked by furosemide, a low [Cl−]i is readjusted by activation of a chloride conductance with the physiologic and pharmacologic properties of ClC-2 (18). In hippocampal neurons, the expression of ClC-2 is correlated with the existence of a low [Cl−]i and a hyperpolarizing GABAergic response. For example, CA1 and CA3 pyramidal neurons, which exhibit a hyperpolarizing inhibitory postsynaptic potential in response to activation of GABAA receptors, express high levels of ClC-2. In contrast, granule cells in the dentate gyrus that are depolarized by GABAergic stimulation do not express this channel (13,14,15,18). Furthermore, ClC-2 mRNA is upregulated postnatally in the rat hippocampus in parallel with the developmental switch of the GABA response from excitatory to inhibitory (19). Finally, experimental gene transfer of ClC-2 into dorsal root ganglion neurons that do not express this channel clamps ECl to the membrane potential and changes the GABA response of these cells from excitatory to inhibitory (20).
Screening for CLCN2 Mutations
We screened the CLCN2 gene in index patients of 46 IGE families linked to chromosome 3q26 using single-strand conformation analysis (SSCA). Direct sequencing of aberrant bands revealed three heterozygous mutations that cosegregate with the IGE trait in three unrelated families, presenting with the typical IGE subtypes of EGMA, CAE, JAE, or JME, respectively (Fig. 20-1). None of these mutations was identified in a total of 360 control chromosomes. No further CLCN2 mutations were found in the index patients by sequencing all coding exons and adjacent splice sites.
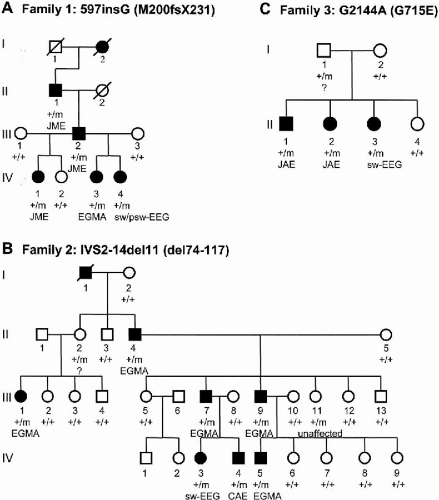 FIG. 20-1. Segregation analysis of three different CLCN2 mutations in families with common IGE subtypes. Individuals carrying one of the mutations are denoted by +/m, whereas the absence of a mutation is denoted by +/+. Filled symbols represent individuals diagnosed with either JME, JAE, CAE, or EGMA. Three individuals were regarded as affected based on their pathologic EEG showing spontaneous generalized spike-wave and/or polyspike-wave discharges. A: Family 1 was ascertained through individual IV-1, who was diagnosed with JME. Segregation analyses showed perfect cosegregation of the mutation and the affection status in this family. B: The index patient in family 2 (IV-4), experienced daily clusters of absence seizures during childhood (CAE). All affected family members carry the mutation, whereas the occurrence of the mutation in the unaffected individual III-11 indicates incomplete penetrance of this mutation. C: Family 3 was ascertained through the index patient II-2 whose epilepsy syndrome was classified as JAE. All affected family members were found to be heterozygous for the mutation. |
The leading IGE syndrome in family 1 was JME, which presented with frequent myoclonic and generalized tonic–clonic seizures (Fig. 20-1A). A single nucleotide insertion in bp-position 597 (597insG) was detected within exon 5 of individual IV:1. The 597insG mutation alters the normal translational reading frame and predicts a premature stop codon (M200fsX231) that severely truncates the protein.
Affected individuals of family 2 experienced rare GTCS on awakening (EGMA), except individual IV:4 who exclusively suffered from absence seizures (CAE) (Fig. 20-1B). An 11-bp deletion in intron 2, IVS2-14del11, was identified in this family, close to the splice acceptor site. This prompted us to search for alternative ClC-2 splice products. RT-PCR using primers located in exons 1 and 5 demonstrated the presence of a novel ClC-2 splice variant lacking exon 3 (delexon3). Skipping of exon 3 leads to an in-frame deletion of 44 amino acid residues (del74-117). Since this splice variant was also found in healthy controls, we established a quantitative competitive RT-PCR assay. In both tested patients carrying the IVS2-14del11 mutation the number of WT mRNA copies was reduced (60% and 66% of the average number found in two controls), whereas the number of delexon3 mRNA copies was increased eight-fold. Accordingly, in the two control subjects, we found a delexon3:WT mRNA ratio of 5:95 and in patients carrying the intronic 11 bp deletion; the ratio was found to be 40:60.
In family 3, two affected individuals were diagnosed with JAE (Fig. 20-1C). We identified a single nucleotide exchange in bp-position 2144 (G2144A) predicting the substitution of glutamate for glycine at position 715 (G715E) located C-terminal to the last transmembrane helix.
Patch Clamp Studies of Expressed CIC-2 Channels
Related posts:
 Myoclonic Status in Nonprogressive Encephalopathies
Severe Myoclonic Epilepsy in Infancy: Dravet Syndrome
Idiopathic Myoclonic-Astatic Epilepsy of Early Childhood—Nosology Based on Electrophysiologic and Long-Term Follow-Up Study of Patients
Eyelid Myoclonia and Absence
Familial Adult Myoclonic Epilepsy (FAME)
Treatment of Myoclonic Epilepsies in Infancy and Early Childhood
Myoclonic Status in Nonprogressive Encephalopathies
Severe Myoclonic Epilepsy in Infancy: Dravet Syndrome
Idiopathic Myoclonic-Astatic Epilepsy of Early Childhood—Nosology Based on Electrophysiologic and Long-Term Follow-Up Study of Patients
Eyelid Myoclonia and Absence
Familial Adult Myoclonic Epilepsy (FAME)
Treatment of Myoclonic Epilepsies in Infancy and Early Childhood
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


