Chapter 1 Clinical overview and phenomenology of movement disorders
Fundamentals
The quotation from William Osler is an apt introduction to this chapter, which offers a description of the various phenomenologies of movement disorders. Movement disorders can be defined as neurologic syndromes in which there is either an excess of movement or a paucity of voluntary and automatic movements, unrelated to weakness or spasticity (Table 1.1). The former are commonly referred to as hyperkinesias (excessive movements), dyskinesias (unnatural movements), and abnormal involuntary movements. In this text, the term dyskinesias is used most often, but all are interchangeable. The five major categories of dyskinesias in alphabetical order are chorea, dystonia, myoclonus, tics, and tremor. Table 1.1 presents the complete list.
| Hypokinesias |
| Hyperkinesias |
Distinguishing between organic and psychogenic causation requires expertise in recognizing the various phenomenologies. Psychogenic movement disorders are covered in Chapter 25.
Those who are interested in keeping up-to-date in the field of movement disorders should refer to the journal Movement Disorders, published 12 times per year by The Movement Disorder Society, Inc. (www.movementdisorders.org). The journal, which is accompanied by two DVDs per year, comes with Movement Disorder Society membership, which is open to all interested medical professionals.
Categories of movements
It is important to note that not all of the hyperkinesias in Table 1.1 are technically classified as abnormal involuntary movements, commonly called AIMS. Movements can be categorized into one of four classes: automatic, voluntary, semivoluntary (also called unvoluntary) (Lang, 1991; Tourette Syndrome Classification Study Group, 1993; Fahn, 2005), and involuntary (Jankovic, 1992). Automatic movements are learned motor behaviors that are performed without conscious effort, e.g., walking an accustomed route, and tapping of the fingers when thinking about something else. Voluntary movements are intentional (planned or self-initiated) or externally triggered (in response to some external stimulus; e.g., turning the head toward a loud noise or withdrawing a hand from a hot plate). Intentional voluntary movements are preceded by the Bereitschaftspotential (or readiness potential), a slow negative potential recorded over the supplementary motor area and contralateral premotor and motor cortex appearing 1–1.5 seconds prior to the movement. The Bereitschaftspotential does not appear with other movements, including the externally triggered voluntary movements (Papa et al., 1991). In some cases, learned voluntary motor skills are incorporated within the repertoire of the movement disorder, such as camouflaging choreic movements or tics by immediately following them with voluntarily executed movements, so-called parakinesias. Semivoluntary (or unvoluntary) movements are induced by an inner sensory stimulus (e.g., need to “stretch” a body part or need to scratch an itch) or by an unwanted feeling or compulsion (e.g., compulsive touching or smelling). Many of the movements occurring as tics or as a response to various sensations (e.g., akathisia and the restless legs syndrome) can be considered unvoluntary because the movements are usually the result of an action to nullify an unwanted, unpleasant sensation. Unvoluntary movements usually are suppressible. Involuntary movements are often non-suppressible (e.g., most tremors and myoclonus), but some can be partially suppressible (e.g., some tremors, chorea, dystonia, stereotypies and some tics) (Koller and Biary, 1989).
The origins of abnormal movements
Many movement disorders are associated with pathologic alterations in the basal ganglia or their connections. The basal ganglia are that group of gray matter nuclei lying deep within the cerebral hemispheres (caudate, putamen, and pallidum), the diencephalon (subthalamic nucleus), the mesencephalon (substantia nigra), and the mesencephalic-pontine junction (pedunculopontine nucleus) (see Chapter 3). There are some exceptions to this general rule. Pathology of the cerebellum or its pathways typically results in impairment of coordination (asynergy, ataxia), misjudgment of distance (dysmetria), and intention tremor. Myoclonus and many forms of tremors do not appear to be related primarily to basal ganglia pathology, and often arise elsewhere in the central nervous system, including cerebral cortex (cortical reflex myoclonus), brainstem (cerebellar outflow tremor, reticular reflex myoclonus, hyperekplexia, and rhythmical brainstem myoclonus such as palatal myoclonus and ocular myoclonus), and spinal cord (rhythmical segmental myoclonus and non-rhythmic propriospinal myoclonus). Moreover, many myoclonic disorders are associated with diseases in which the cerebellum is involved, such as those causing the Ramsay Hunt syndrome of progressive myoclonic ataxia (see Chapter 20). The peripheral nervous system can give rise to abnormal movements also, such as the painful legs–moving toes syndrome (Marsden, 1994). It is not known for certain which part of the brain is associated with tics, although the basal ganglia and the limbic structures have been implicated. Certain localizations within the basal ganglia are classically associated with specific movement disorders: substantia nigra with bradykinesia and rest tremor; subthalamic nucleus with ballism; caudate nucleus with chorea; and putamen with dystonia.
Historical perspective
The neurologic literature contains a number of seminal papers, reviews and books that emphasized and established movement disorders as associated with the basal ganglia pathology (Alzheimer, 1911; Fischer, 1911; Wilson, 1912; Hunt, 1917; Vogt and Vogt, 1920; Jakob, 1923; Putnam et al., 1940; Denny-Brown, 1962; Martin, 1967).
An historical perspective of movement disorders can be gained by listing the dates when the various clinical entities were first introduced (Table 1.2).
Table 1.2 Some notable historical descriptions of movement disorders
| Year | Source | Entity |
|---|---|---|
| Bible | Reference to tremor in the aged | |
| Trembling associated with fear and strong emotion | ||
| 1567 | Paracelsus | Mercury-induced tremor |
| 1652 | Tulpius | Spasmodic torticollis |
| 1685 | Willis | Restless legs syndrome |
| 1686 | Sydenham | Sydenham chorea |
| 1817 | Parkinson | Parkinson disease |
| 1825 | Itard | Tourette syndrome |
| 1830 | Bell | Writer’s cramp |
| 1837 | Couper | Manganese-induced parkinsonism |
| 1848 | Grisolle | Primary writing tremor |
| 1871 | Hammond | Athetosis |
| 1871 | Traube | Spastic dysphonia |
| 1871 | Steinthal | Apraxia |
| 1872 | Huntington | Huntington disease |
| 1872 | Mitchell | Jumpy stumps |
| 1874 | Kahlbaum | Catatonia |
| 1878 | Beard | Jumpers |
| 1881 | Friedreich | Myoclonus |
| 1885 | Gilles de la Tourette | Tourette syndrome |
| 1885 | Gowers | Paroxysmal kinesigenic choreoathetosis |
| 1886 | Spencer | Palatal myoclonus |
| 1887 | Dana | Hereditary tremor |
| 1887 | Wood | Cranial dystonia |
| 1889 | Benedikt | Benedikt syndrome |
| 1891 | Unverricht | Progressive myoclonus epilepsy (Unverricht–Lundborg disease) |
| 1895 | Schultze | Myokymia |
| 1900 | Dejerine/Thomas | Olivopontocerebellar atrophy |
| 1900 | Liepmann | Apraxia |
| 1901 | Haskovec | Akathisia |
| 1903 | Batten | Neuronal ceroid lipofuscinosis |
| 1904 | Holmes | Midbrain (“rubral”) tremor |
| 1908 | Schwalbe | Familial dystonia |
| 1910 | Meige | Oromandibular dystonia |
| 1911 | Oppenheim | Dystonia musculorum deformans |
| 1911 | Lafora | Lafora disease |
| 1912 | Wilson | Wilson disease |
| 1914 | Lewy | Lewy bodies in Parkinson disease |
| 1916 | Henneberg | Cataplexy |
| 1917 | Hunt | Progressive pallidal atrophy |
| 1920 | Creutzfeldt | Creutzfeldt–Jakob disease |
| 1921 | Jakob | Creutzfeldt–Jakob disease |
| 1921 | Hunt | Dyssynergia cerebellaris myoclonica (Ramsay Hunt syndrome) |
| 1922 | Hallervorden/Spatz | Pantothenate kinase deficiency (neurodegenerative disorder with brain iron deposition-1) |
| 1923 | Sicard | Akathisia |
| 1924 | Fleischhacker | Striatonigral degeneration |
| 1926 | Davidenkow | Myoclonic dystonia |
| 1927 | Goldsmith | Hereditary chin quivering |
| 1927 | Orzechowski | Opsoclonus |
| 1931 | Herz | Myorhythmia |
| 1931 | Guillain/Mollaret | Palato-pharyngo-laryngo-oculo-diaphragmatic myoclonus |
| 1932 | De Lisi | Hypnic jerks |
| 1933 | Spiller | Fear of falling |
| 1933 | Scherer | Striatonigral degeneration |
| 1940 | Mount/Reback | Paroxysmal nonkinesigenic dyskinesia (paroxysmal dystonic choreoathetosis) |
| 1941 | Louis-Bar | Ataxia-telangiectasia |
| 1943 | Kanner | Autism |
| 1944 | Asperger | Autism |
| 1946 | Titeca/van Bogaert | Dentatorubral-pallidoluysian degeneration |
| 1949 | Alexander | Alexander disease |
| 1953 | Adams/Foley | Asterixis |
| 1953 | Symonds | Nocturnal myoclonus (periodic movements in sleep) |
| 1954 | Davison | Pallido-pyramidal syndrome (PARK15) |
| 1956 | Moersch/Woltman | Stiff-person syndrome |
| 1957 | Schonecker | Tardive dyskinesia |
| 1958 | Kirstein/Silfverskiold | Startle disease (hyperekplexia) |
| 1958 | Smith et al. | Dentatorubral-pallidoluysian degeneration |
| 1958 | Monrad-Krohn/Refsum | Myorhythmia |
| 1959 | Paulson | Acute dystonic reaction |
| 1960 | Ekbom | Restless legs |
| 1960 | Shy/Drager | Dysautonomia with parkinsonism (multiple system atrophy) |
| 1961 | Hirano et al. | Parkinsonism-dementia complex of Guam |
| 1961 | Andermann et al. | Facial myokymia |
| 1961 | Isaacs | Neuromyotonia, Isaacs syndrome |
| 1962 | Kinsbourne | Opsoclonus-myoclonus |
| 1963 | Lance/Adams | Posthypoxic action myoclonus |
| 1964 | Adams et al. | Striatonigral degeneration |
| 1964 | Steele et al. | Progressive supranuclear palsy |
| 1964 | Levine | Neuroacanthocytosis |
| 1964 | Kinsbourne | Sandifer syndrome |
| 1964 | Lesch/Nyhan | Lesch–Nyhan syndrome |
| 1965 | Hakim/Adams | Normal pressure hydrocephalus |
| 1965 | Goldstein/Cogan | Apraxia of lid opening |
| 1966 | Suhren et al. | Hyperekplexia |
| 1966 | Rett | Rett syndrome |
| 1967 | Haerer et al. | Hereditary nonprogressive chorea |
| 1968 | Rebeiz et al. | Cortical-basal ganglionic degeneration |
| 1968 | Delay/Denniker | Neuroleptic malignant syndrome |
| 1969 | Horner/Jackson | Hypnogenic paroxysmal dyskinesias |
| 1969 | Graham/Oppenheimer | Multiple system atrophy |
| 1970 | Spiro | Minipolymyoclonus |
| 1970 | Ritchie | Jumpy stumps |
| 1971 | Spillane et al. | Painful legs and moving toes |
| 1975 | Perry et al. | Familial parkinsonism with hypoventilation and mental depression |
| 1976 | Segawa et al. | Dopa-responsive dystonia |
| 1976 | Allen/Knopp | Dopa-responsive dystonia |
| 1977 | Hallett et al. | Reticular myoclonus |
| 1978 | Satoyoshi | Satoyoshi syndrome |
| 1978 | Fahn | Tardive akathisia |
| 1979 | Hallett et al. | Cortical myoclonus |
| 1979 | Rothwell et al. | Primary writing tremor |
| 1980 | Fukuhara et al. | Myoclonus epilepsy associated with ragged red fibers (MERFF) |
| 1980 | Coleman et al. | Periodic movements in sleep |
| 1981 | Fahn/Singh | Oscillatory myoclonus |
| 1981 | Lugaresi/Cirignotta | Hypnogenic paroxysmal dystonia |
| 1982 | Burke et al. | Tardive dystonia |
| 1983 | Langston et al. | MPTP-induced parkinsonism |
| 1984 | Heilman | Orthostatic tremor |
| 1985 | Aronson | Breathy dysphonia |
| 1986 | Bressman et al. | Biotin-responsive myoclonus |
| 1986 | Schenck et al. | REM sleep behavior disorder |
| 1986 | Schwartz et al. | Oculomasticatory myorhythmia |
| 1987 | Tominaga et al. | Tardive myoclonus |
| 1987 | Little/Jankovic | Tardive myoclonus |
| 1990 | Iliceto et al. | Abdominal dyskinesias |
| 1990 | Ikeda et al. | Cortical tremor/myoclonus |
| 1991c | Brown et al. | Propriospinal myoclonus |
| 1991 | Hymas et al. | Obsessional slowness |
| 1991 | De Vivo et al. | GLUT1 deficiency syndrome |
| 1992 | Stacy/Jankovic | Tardive tremor |
| 1993 | Bhatia et al. | Causalgia-dystonia |
| 1993 | Atchison et al. | Primary freezing gait |
| 1993 | Achiron et al. | Primary freezing gait |
| 2002 | Namekawa et al. | Adult-onset Alexander disease |
| 2002 | Okamoto et al. | Adult-onset Alexander disease |
Other important dates in the history of movement disorders are 1912, the coining of the term “extrapyramidal” by Wilson; 1985, the founding of the Movement Disorder Society, and 1986, the publication of the first issue of the journal, Movement Disorders.
Epidemiology
Movement disorders are common neurologic problems, and epidemiological studies are available for some of them (Table 1.3). There have been several studies for Parkinson disease (PD), and these have been carried out in several countries (Tanner, 1994; de Lau and Breteler, 2006). Table 1.3 lists the prevalence rates of some movement disorders based on studies in the United States. The frequency of different types of movement disorders seen in the two specialty clinics at Columbia University and Baylor College of Medicine are presented in Table 1.4. More detailed information is provided in the relevant chapters for specific diseases.
Table 1.3 Prevalence of movement disorders
| Disorder | Rate per 100 000 | Reference |
|---|---|---|
| Restless legs | 9800* | Rothdach et al. (2000) |
| Essential tremor | 415 | Haerer et al. (1982) |
| Parkinson disease | 187† | Kurland (1958) |
| Tourette syndrome | 29–1052 | Caine et al. (1988), Comings et al. (1990) |
| 2990 | Mason et al. (1998) | |
| Primary torsion dystonia | 33 | Nutt et al. (1988) |
| Hemifacial spasm | 7.4–14.5 | Auger and Whisnant (1990) |
| Blepharospasm | 13.3 | Defazio et al. (2001) |
| Hereditary ataxia | 6 | Schoenberg (1978) |
| Huntington disease | 2–12 | Harper (1992), Kokmen et al. (1994) |
| Wilson disease | 3 | Reilly et al. (1993) |
| Progressive supranuclear palsy | 2 | Golbe (1994) |
| 2.4 | Nath et al. (2001) | |
| 6.4 | Schrag et al. (1999) | |
| Multiple system atrophy | 4.4 | Schrag et al. (1999) |
Rates are given per 100 000 population. *For restless legs, the rate cited is in a population 65–83 years of age. †For Parkinson disease, the rate is 347 per 100 000 for ages over 39 years (Schoenberg et al., 1985).
Table 1.4 The prevalence of movement disorders encountered in two large movement disorder clinics
| Movement disorder | Number of patients | Percent |
|---|---|---|
| Parkinsonism | 15 107 | 35.3 |
| Parkinson disease | 10 182 | |
| Progressive supranuclear palsy | 750 | |
| Multiple system atrophy | 841 | |
| Cortical-basal ganglionic degeneration | 297 | |
| Vascular | 867 | |
| Drug-induced | 327 | |
| Hemiparkinsonism–hemiatrophy | 116 | |
| Gait disorder | 329 | |
| Other | 1 308 | |
| Dystonia | 10 394 | 24.3 |
| Primary dystonia | 7 784 | |
| Focal | (59%) | |
| Segmental | (29%) | |
| Generalized | (12%) | |
| Secondary dystonia | 6 610 | |
| Hemidystonia | 279 | |
| Tardive | 595 | |
| Other | 1 737 | |
| Tremor | 6 754 | 15.8 |
| Essential tremor | 2 818 | |
| Cerebellar | 205 | |
| Midbrain (“rubral”) | 88 | |
| Primary writing | 114 | |
| Orthostatic | 82 | |
| Other | 1 035 | |
| Tics (Tourette syndrome) | 2 753 | 6.4 |
| Chorea | 1 225 | 2.9 |
| Huntington disease | 690 | |
| Hemiballism | 123 | |
| Other | 412 | |
| Tardive syndromes | 1 253 | 2.9 |
| Myoclonus | 1 020 | 2.4 |
| Hemifacial spasm | 693 | 1.6 |
| Ataxia | 764 | 1.9 |
| Paroxysmal dyskinesias | 474 | 1.1 |
| Stereotypies (other than TD) | 246 | 0.6 |
| Restless legs syndrome | 807 | 1.9 |
| Stiff-person syndrome | 70 | 0.2 |
| Psychogenic movement disorder | 1 268 | 3.0 |
| Grand total | 42 826 | 100 |
The above data were obtained from the combined databases of the Movement Disorder Clinics at Columbia University Medical Center (New York City) and Baylor College of Medicine (Houston) for patients encountered through April 2009. Because some patients might have more than one type of movement disorder (such as a combination of essential tremor and Parkinson disease), they would be listed more than once. Therefore, the figures in the table represent the types of movement disorder phenomenology encountered in two large clinics, rather than the exact number of patients.
Genetics
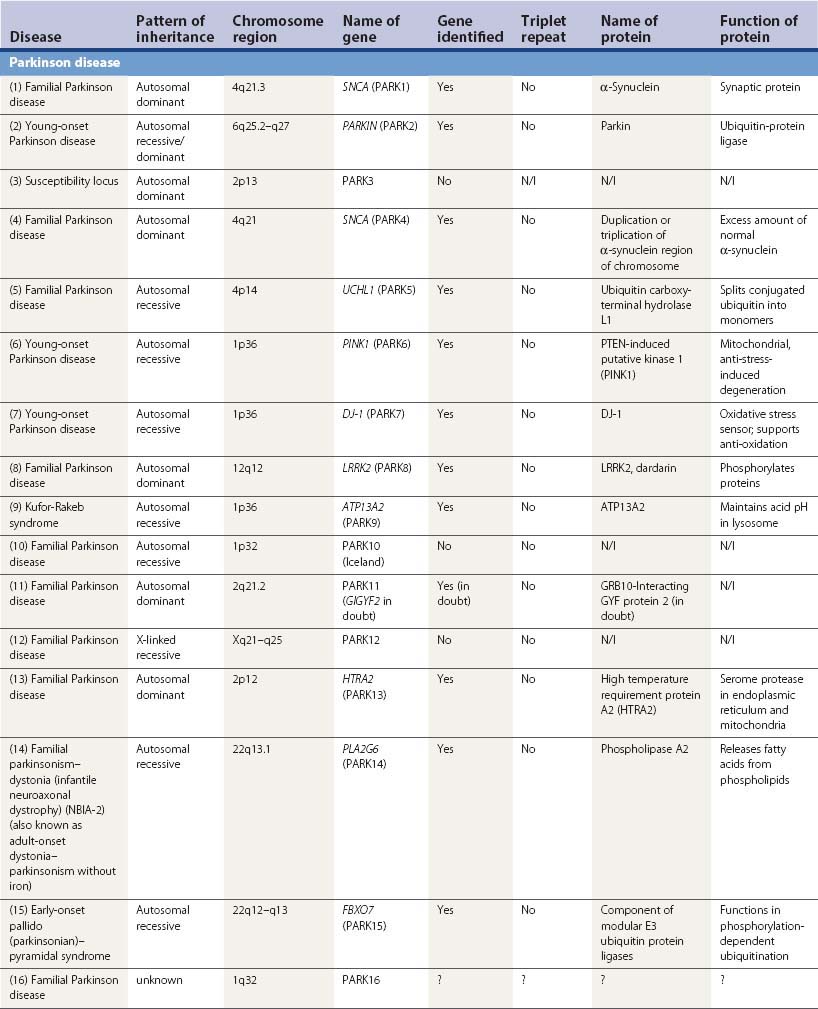
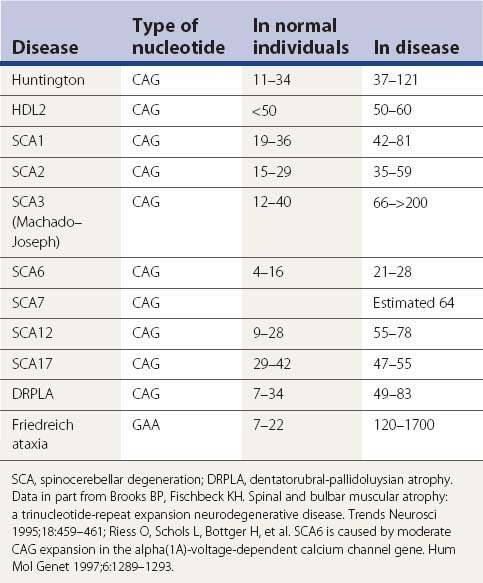
A large number of movement disorders are genetic in etiology, and many of the diseases have now been mapped to specific regions of the genome, and some have even been localized to a specific gene (Table 1.5). For example, ten genetic loci have so far been identified with Parkinson disease (PD) or variants of classic PD (PARK4 is a triplication of the normal α-synuclein gene, for which mutations are listed as PARK1). Several genetic loci of movement disorders have been identified with a specific gene and protein. A comprehensive list of movement disorders whose genes have been mapped or identified are listed in Table 1.5. A detailed chapter (Harris and Fahn, 2003) and an entire book (Pulst, 2003) have been published specifically related to movement disorder genetics. Several inherited movement disorders are due to expanded repeats of the trinucleotide cytosine-adenosine-guanosine (CAG), and Friedreich ataxia is due to the expanded trinucleotide repeat of guanosine-adenosine-adenosine (GAA). Normal individuals contain an acceptable number of these trinucleotide repeats in their genes, but these triplicate repeats are unstable, and when expanded, lead to disease (Table 1.6). Neurogenetics is one of the fastest moving research areas in neurology, so the list in Table 1.5 keeps expanding rapidly.
Quantitative assessments
The assessment of severity of disease is a process that is carried out by all clinicians when evaluating a patient. Quantifying the severity provides the means of determining the progression of the disorder and the effect of intervention by pharmacologic or surgical approaches. Many mechanical and electronic devices, including accelerometers, can quantitate specific signs, such as tremor, rigidity, and bradykinesia. These have been developed by physicians and engineers over at least 80 years (Lewy, 1923; Carmichael and Green, 1928), and newer computerized devices continue to be conceived and developed (Larsen et al., 1983; Tryon, 1984; Potvin and Tourtelotte, 1985; Cohen et al., 2003). The advantages of mechanical and electronic measurements are objectivity, consistency, uniformity among different investigators, and rapidity of database storage and analysis. However, these measurements might not be as sensitive as more subjective clinical measurements. In one study comparing objective measurements of reaction and movement times with clinical evaluations, Ward and his colleagues (1983) found the latter to be more sensitive.
The mechanical and electronic methods of measurement have other disadvantages. Instrumentation can usually measure only a single sign, at a single point in time, and in a single part of the body. Disorders such as parkinsonism encompass a wide range of motor abnormalities, as well as behavioral features. Clinical measurements can cover a wider range of the parkinsonian spectrum of impairments, and have the advantage of being carried out at the bedside or in the office or clinic at the time the patient is being examined by the physician. Equally important, clinical assessment can evaluate disability in terms of activities of daily living (ADL), and the one developed by England and Schwab (1956) and modified slightly (Fahn and Elton, 1987) has proven highly useful.
A number of clinical rating scales have been proposed (e.g., see Marsden and Schachter, 1981). Several that are now considered standards and are in wide use can be recommended: the Unified Parkinson’s Disease Rating Scale (UPDRS) (Fahn and Elton, 1987) is the standard scale for rating severity of signs and symptoms; a videotaped demonstration of the assigned ratings has been published (Goetz et al., 1995). A modification of the UPDRS by the Movement Disorder Society is underway (Goetz et al., 2007) and will be known as the MDS-UPDRS. Other standard scales for PD and its complications are the Schwab and England Activities of Daily Living scale for parkinsonism (Schwab and England, 1969) as modified (Fahn and Elton, 1987); the Hoehn and Yahr Parkinson Disease Staging Scale (Hoehn and Yahr, 1967) as modified (Fahn and Elton, 1987); the Goetz dopa dyskinesia severity scale (Goetz et al., 1994); the Lang–Fahn dopa dyskinesia ADL scale (Parkinson Study Group, 2001); the Parkinson psychosis scale (Friedberg et al., 1998); the daily diary to record fluctuations and dyskinesias (Hauser et al., 2004); the core assessment program for intracerebral transplantation (Langston et al., 1992); the PSP Rating Scale (Golbe and Ohman-Strickland, 2007); the Fahn–Marsden Dystonia Rating Scale (Burke et al., 1985); the Unified Dystonia Rating Scale (Comella et al., 2003); the Fahn–Tolosa clinical rating scale for tremor (Fahn et al., 1993); the Bain tremor scale (Bain et al., 1993); and the Unified Huntington’s Disease Rating Scale, which also has a published videotaped demonstration of assigned ratings (Huntington Study Group, 1996).
Differential diagnosis of hypokinesias
For a list of hypokinesias, refer to Table 1.1.
Akinesia/Bradykinesia
Akinesia, bradykinesia, and hypokinesia literally mean “absence,” “slowness,” and “decreased amplitude” of movement, respectively. The three terms are commonly grouped together for convenience and usually referred to under the term of bradykinesia. These phenomena are a prominent and most important feature of parkinsonism, and are often considered a sine qua non for parkinsonism. Although akinesia means “lack of movement,” the label is often used to indicate a very severe form of bradykinesia (Video 1.1). Bradykinesia is mild in early PD, and becomes more severe as the disease worsens; similarly in other forms of parkinsonism. A discussion of the phenomenology of akinesia/bradykinesia requires a brief description of the clinical features of parkinsonism. A fuller discussion is presented in Chapter 4. 
Parkinsonism is a neurologic syndrome manifested by any combination of six independent, non-overlapping cardinal motor features: tremor-at-rest, bradykinesia, rigidity, flexed posture, freezing, and loss of postural reflexes (Table 1.7). At least two of these six cardinal features should be present before the diagnosis of parkinsonism is made, one of them being tremor-at-rest or bradykinesia. There are many causes of parkinsonism; they can be divided into four major categories: primary, secondary, parkinsonism-plus syndromes, and heredodegenerative disorders (Table 1.8). Primary parkinsonism (Parkinson disease) is a progressive disorder of unknown etiology or of a known gene defect, and the diagnosis is usually made by excluding other known causes of parkinsonism (Fahn, 1992). The complete classification of parkinsonian disorders is presented in Chapter 4. The specific diagnosis of the type of parkinsonism depends on details of the clinical history, the neurologic examination, and laboratory tests.
Table 1.7 Cardinal features of parkinsonism
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
