CHAPTER 331 Clinical Pathophysiology of Traumatic Brain Injury
The initial mechanical insult of traumatic brain injury (TBI) results in tissue deformation that causes damage to neurons, glia, axons, and blood vessels. This is followed by a more delayed phase of injury, which is mediated by intracellular and extracellular biologic pathways and can be present for minutes, hours, days, and even weeks after the primary insult.1–6 During this phase, many patients experience superimposed secondary insults such as hypoxia, hypotension, cerebral swelling, and the consequences of increased intracranial pressure (ICP). These secondary insults7 further exacerbate TBI and have a profound negative effect on patient outcome. An understanding of the sequelae of TBI and secondary insults is paramount for managing head injury; all current neurosurgical and neurocritical care interventions target the delayed effects of secondary injuries and secondary insults.
In a recent report from the International Mission for Prognosis and Clinical Trial (IMPACT) database, hypoxia and hypotension were present on admission in 20% and 18% of TBI patients.8 These insults can be quantified using a system described by Miller and colleagues.9,10 In this study, 124 patients with mild, moderate, or severe head injury were monitored for the following secondary insults: elevated ICP, reduced cerebral perfusion pressure (CPP), hypotension, hypoxia, jugular venous oxygen desaturation, and presence of pyrexia, tachycardia, and bradycardia. Overall, 90% of patients experienced one or more secondary insults, with 50% sustaining an insult of highest severity grade. Secondary insults were most common in the severely injured group (67 of 68) and occurred less frequently in moderate (7 of 36) and mild (3 of 20) injury groups. The authors found that 50% of patients sustained a secondary insult during transport within the hospital, and repeat secondary insults were common even during intensive care management.7 Five insults consistently correlated with poor outcome in this study: arterial hypotension, reduced CPP, elevated ICP, hypoxemia, and pyrexia.10
Although treating secondary injury mechanisms and preventing secondary insults can be challenging, mortality after severe TBI has significantly improved from 39% in 1984 to 27% in 1996.11 This is due, in large part, to increased recognition of the mechanisms underlying secondary brain injury and optimization of cerebral perfusion and oxygenation to avoid secondary brain insults. Significant advances have been made in prehospital care, resuscitation, and rapid radiologic diagnosis,12 and there is increasing emphasis placed on early and aggressive surgical and medical treatment of intracranial mass lesions, raised ICP, and decreased CPP.13–17
No particular treatment has been shown to improve outcome in a randomized, controlled clinical trial, partly owing to ethical concerns.18–22 However, many authors have shown that protocol-driven therapy for TBI significantly improves patient outcome,23–26 especially when aggressive care27 is provided in specialized neurocritical care settings.28 As a result, various TBI guidelines have been developed and updated to guide patient care. These address topics such as prehospital management,29 penetrating TBI,13 surgical management,30 and critical care management of infants, children, and adolescents31 as well as adults.32 Further improvements in management and outcome of TBI patients will likely depend on improved understanding of the pathophysiology of this extremely complex disease. It is important to note, however, that none of these protocols have proved effective in a randomized clinical trial.
In this chapter, we discuss the pathogenesis of closed head injury and the effect of trauma on cerebral metabolism and circulation. Basic concepts of therapeutic intervention, as they pertain to these processes, are reviewed. Penetrating head injury is described in Chapter 331, and specific medical and surgical management of TBI is discussed in Chapter 326, Chapter 327 to Chapter 328.
Mechanisms of Brain Injury
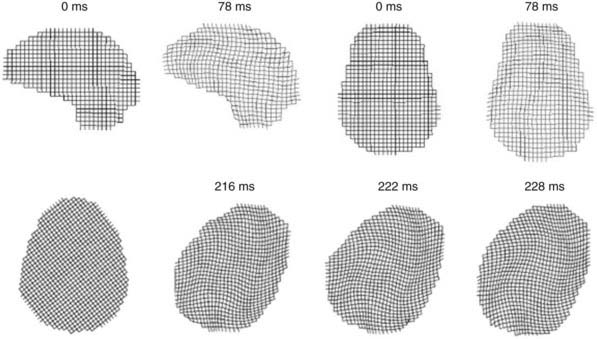
The biomechanics of closed head injury have been extensively described in animals,33–38 human cadavers,39–42 and experimental models of the skull and brain.13,36,43–45 In 1943, Holbourn44 demonstrated the effects of rotational forces on gel housed within a human skull, and 3 years later, Pudenz and Shelden46 visually recorded this phenomenon in a monkey skull replaced with a transparent plastic top. High-speed filming of gel-filled skulls36,47 and high-speed biplanar radiography of cadaveric brains48 have shed additional light on brain deformation after head injury. More recently, computational models49 and magnetic resonance imaging (MRI) techniques have been adapted to study the biomechanical properties of head injury. For example, Bayly and associates have studied the effects of mild linear50 and angular51 head acceleration on brain deformation in healthy volunteers (Fig. 331-1). Their data suggest that mechanical responses are mediated by divisions between brain regions (e.g., central sulcus), dural reflections (e.g., falx cerebri, tentorium cerebelli), and tethering of the brain at the sella and suprasellar regions.
In 1966, Goldsmith defined three physical processes causing head injury52: collision of the head with a solid object at an appreciable velocity, an impulsive load producing sudden motion of the head without significant physical contact, and a static or quasistatic load compressing the head with gradual force. Collision typically results in brain injury through a combination of contact and inertial forces,53 whereas impulsive forces cause inertial loading to the head. Although mild injuries such as concussion may result from this process, impulsive forces typically occur in conjunction with a collision or impact mechanism.54 A static or quasistatic load also involves a contact force, but the speed of impact is minimal or zero. In this scenario, the contribution of inertial forces is negligible, and damage is caused by gradually increasing contact forces trapping the head against a rigid structure.
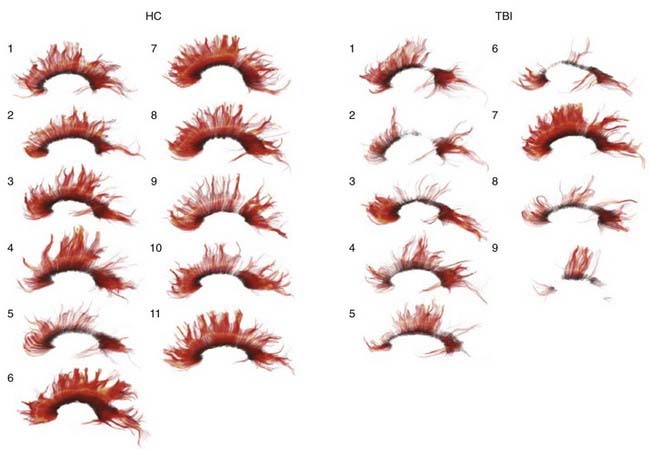
Contact forces typically result in focal injuries such as coup contusions and skull fractures. Inertial loading forces that are primarily translational also result in focal injuries, such as contusions and subdural hematomas, whereas rotational acceleration-deceleration injuries are more likely to result in diffuse injuries ranging from concussion to diffuse axonal injury (DAI). Rotational injuries are particularly concerning because they cause injury to both the cortical surface and deep brain structures. Recently, diffusion tensor imaging (DTI) techniques have been used to better visualize the distribution and severity of white matter fiber injuries after DAI (Fig. 331-2).55,56
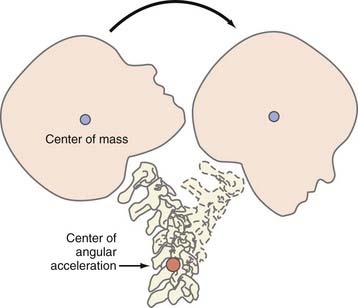
Angular acceleration represents a combination of translational and rotational acceleration and is the most common form of inertial injury. Because of the biomechanical properties of the head and neck, TBI often results in deflection of the head and neck around the middle or lower cervical spine (the center of angular movement; Fig. 331-3). The resultant magnitude of rotation that occurs with this injury depends on the distance between the center of gravity and the center of angulation: the smaller the distance, the larger the rotational component of angulation.57
The extent of injury after TBI is also determined, in large part, by the magnitude and duration of the insult mechanism. In an experimental model of angular acceleration, the influence of duration of the acceleration force, the time to peak acceleration, and the magnitude of acceleration were tested; a threshold value was established below which the impact resulted in concussion rather than a subdural hematoma.33 The study also found that a low magnitude of acceleration with a long duration results in DAI, owing to propagation of the forces deep within the brain. In contrast, a brief, high-velocity impact often results in tearing of superficially located bridging veins and pial vessels, causing subdural hematoma. The former mechanism is typically seen with motor vehicle collisions, whereas the latter occurs in falls or assaults in which the head strikes a broad, hard surface, and inertial loading is the predominant mechanism.33,58
Classification of Head Injury
Clinical condition and level of consciousness after TBI are typically described using the Glasgow Coma Scale (GCS; Table 331-1). This scale has been universally adopted for grading the clinical severity of head injuries and other pathologies that impair consciousness.59 Before the computed tomography (CT) era, GCS score was used to identify patients who should undergo contrast studies to detect intracranial collections of blood. In the modern era, however, GCS plays an important role in categorizing injury severity, allowing for standardized determination of clinical neurological status and detecting episodes of neurological deterioration. It must be noted, however, that the utility of GCS is somewhat limited with modern-day therapies. For example, most patients arrive to the hospital by ambulance unresponsive because of sedation and neuromuscular blockade. In these patients, an accurate postresuscitation score cannot be determined until pharmacologic agents are actively antagonized or metabolized. Similar challenges are faced during intensive care unit (ICU) management because many ICP-lowering therapies render patients heavily sedated, paralyzed, or both. Furthermore, intubation and concomitant injuries resulting in orbital swelling can significantly interfere with accurate eye opening and verbal scoring. In a study by Gale and associates,60 50% of patients could not be assigned an accurate GCS score because of these confounding variables. An alternative scale for scoring clinical condition is the head injury severity scale (Table 331-2), but it faces similar challenges because it is largely based on the GCS score.61
TABLE 331-1 Scoring System of the Glasgow Coma Scale
| TEST | SCORE |
|---|---|
| Eye Opening (E) | |
| Spontaneous | 4 |
| Open to voice | 3 |
| Open to pain | 2 |
| None | 1 |
| Best Motor Response (M) | |
| Following commands | 6 |
| Localizing to painful stimulus | 5 |
| Flexion-withdrawal to painful stimulus | 4 |
| Flexor/decorticate posturing to painful stimulus | 3 |
| Extensor/decerebrate posturing to painful stimulus | 2 |
| None | 1 |
| Best Verbal Response (V) | |
| Oriented conversation | 5 |
| Confused/disoriented conversation | 4 |
| Inappropriate words | 3 |
| Incomprehensible sounds | 2 |
| None* | 1 |
| Maximum Score (E + M + V) | 15 |
* Patients who are intubated receive a verbal score of “T,” and the scale is adjusted to 3T-11T.
TABLE 331-2 Head Injury Severity Scale
| INJURY CATEGORY | GLASGOW COMA SCALE SCORE |
|---|---|
| Minimal | 15, no loss of consciousness (LOC) or amnesia |
| Mild | 14-15 with amnesia, brief LOC, or impaired alertness |
| Moderate | 9-13, LOC ≥ 5 minutes or focal neurological deficit |
| Severe | 5-8 |
| Critical | 3-4 |
Adapted from Stein S: Classification of head injury. In Narayan RK, Wilberger JE, Povlishock JT, eds: Neurotrauma. New York: McGraw-Hill; 1996:31-41.
Because of the widespread use of GCS by paramedics, emergency physicians, and surgical and critical care colleagues, it is important for the practicing neurosurgeon to understand the anatomic correlation of the GCS score. One persistent misconception about the GCS is that loss of a flexor response (loss of decorticate rigidity and posturing) represents a lesion that affects descending rubrospinal tracts originating from the upper midbrain, and that the extensor response (decerebrate rigidity and posturing) represents output primarily from the pons and medulla. Although this anatomic relationship was classically described in animal studies from the Sherrington preparations,62,63 more recent studies suggest that these responses in human TBI do not strictly correspond to anatomic lesions. Instead, Greenberg and colleagues found that GCS can be related to the severity of injury at any site in the brain; absent motor response or decerebrate motor posturing can occur with severe, purely cortical, or hemispheric lesions.64
TBI can also be classified anatomically into focal or diffuse injury patterns (Table 331-3). By using CT and MRI to describe injury patterns, anatomic classifications can be used to identify patients at increased risk for secondary insults, delayed neurological worsening, and poor clinical outcome. In 1991, the National Institutes of Health (NIH) Trauma Coma Data Bank (TCDB) introduced a classification system for head injury based on initial CT scan findings (Table 331-4).65 Recognized as the Marshall score, this classification system has been used to design clinical trials, guide patient management, and predict outcome based on radiographic criteria. Recently, Marshall, Maas, and colleagues66 proposed a modified version of the Marshall score (termed the Rotterdam score) to account for additional radiographic criteria that more accurately predict survival from head injury (Table 331-5). These systems describe a strong correlation between CT scan findings (e.g., compression of the basal cisterns, presence of subarachnoid hemorrhage, midline shift) and clinical course, mortality, and functional outcome after TBI.67 Because determining an accurate postresuscitation GCS score is extremely difficult in the current era, it is likely that radiographic scoring systems will play a larger role in predicting outcome and directing care in the acute period following TBI.
TABLE 331-3 Focal Compared with Diffuse Injury
| FOCAL INJURIES | DIFFUSE INJURIES |
|---|---|
| Contusions | Concussion |
| Fracture | Diffuse axonal injury |
| Coup | Moderate |
| Contrecoup | Severe |
| Herniation | |
| Intermediate | |
| Gliding | |
| Hematomas | |
| Epidural | |
| Subdural | |
| Intracerebral |
TABLE 331-4 Classification of Head Injury Based on Initial Computed Tomography Findings: Marshall Score
| CATEGORY | DEFINITION |
|---|---|
| Diffuse injury I | No visible intracranial pathology |
| Diffuse injury II | Cisterns present with midline shift 0-5 mm and/or: |
| Lesion densities present | |
| No high- or mixed-density lesion >25 mL | |
| May include bone fragments and foreign bodies | |
| Diffuse injury III (swelling) | Cisterns compressed or absent with midline shift 0-5 mm; no high- or mixed-density lesion >25mL |
| Diffuse injury IV | Midline shift >5 mm; no high- or mixed-density lesion >25 mL |
| Evacuated mass lesion | Any lesion surgically evacuated |
| Nonevacuated mass lesion | High- or mixed-density lesion >25 mL, not surgically evacuated |
From Marshall L, Bowers S, Klauber M. A new classification of head injury based on computerized tomography. J Neurosurg. 1991;75:514-520.
TABLE 331-5 Classification of Head Injury Based on Initial Computed Tomography Findings: Rotterdam Score
| PREDICTOR VALUE | SCORE |
|---|---|
| Basal Cisterns | |
| Normal | 0 |
| Compressed | 1 |
| Absent | 2 |
| Midline Shift | |
| ≤5 mm | 0 |
| >5 mm | 1 |
| Epidural Mass Lesion | |
| Present | 0 |
| Absent | 1 |
| IVH or Traumatic SAH | |
| Absent | 0 |
| Present | 1 |
| Sum score | +1 |
| Total | 1-6 |
IVH, intraventricular hemorrhage; SAH, subarachnoid hemorrhage.
From Maas A, Hukkelhoven C, Marshall L, Steyerberg E. Prediction of outcome in traumatic brain injury with computed tomographic characteristics: a comparison between the computed tomographic classification and combinations of computed tomographic predictors. Neurosurgery. 57:1173-82, 2005.
Focal Brain Injury
Focal brain injuries typically result in contusions and traumatic intracranial hematomas (see Table 331-3).
Brain Contusion
Brain contusions represent focal regions of subpial hemorrhage and swelling and are present in 31% of patients on initial CT scan.68,69 When the overlying pia mater is compromised, the lesion is termed a laceration, although this distinction is typically not clear. Contusions are most common in regions that contact bony surfaces in the cranial vault during trauma: frontal and temporal poles, orbitofrontal gyri, perisylvian cortices, and inferolateral temporal lobe surfaces.
Contusions can be characterized by mechanism, anatomic location, or adjacent injuries. For example, fracture contusions result from direct contact injuries and occur immediately adjacent to a skull fracture.70 Coup contusions refer to those that occur at the site of impact in the absence of a fracture, whereas contrecoup contusions are those that are diametrically opposite to the point of impact. Gliding contusions are focal hemorrhages involving the cortex and adjacent white matter of the superior margins of the cerebral hemispheres; they are due to rotational mechanisms rather than contact forces.71 Intermediary contusions are lesions that affect deep brain structures, such as the corpus callosum, basal ganglia, hypothalamus, and brainstem.71 Herniation contusions can occur in areas where the medial parts of the temporal lobe contact the tentorial edge (i.e., uncal herniation) or where the cerebellar tonsils contact the foramen magnum (i.e., tonsillar herniation).
Contusions typically result in varying degrees of neurological deficits depending on the area involved. Occasionally, contusions can cause significant mass effect owing to surrounding edema or hemorrhagic progression to an intracerebral hematoma (discussed later). Contusions also represent a significant source of secondary injury to adjacent tissue through release of neurotransmitter and local biochemical changes.5 Adams and colleagues proposed a method for quantifying cerebral contusions (contusion index) caused by nonmissile head injury.72,73 They found that contusions are most severe in the frontal and temporal lobes and do not consistently correlate with injury mechanism. Contusions are more severe when associated with a skull fracture, less severe in patients with DAI, and more severe in patients who do not experience a lucid interval.
Traumatic Intracranial Hematoma
Intracranial hematomas represent mass lesions that are potential therapeutic targets of surgical intervention (as opposed to most contusions). They are more common in patients with skull fractures (Table 331-6). About half of patients with severe TBI and skull fracture have a sizeable intracranial hematoma on initial head CT.10,74 The three major types of traumatic intracranial hematomas are distinguished by their location relative to the meninges: epidural, subdural, and intracerebral.
TABLE 331-6 Relationship between Skull Fracture and Intracranial Hematoma in Patients with Severe, Moderate, and Minor Head Injury

Epidural Hematoma
Epidural hematomas (EDHs) occur in 1% to 2% of TBI patients admitted to the hospital75 and account for 5% to 15% of fatal head injuries.76 They are most common in patients younger than 50 years, although they do occur in all age groups.77–80 In adults, EDH is far less common than subdural or intracerebral hemorrhage. In pediatric patients, however, EDH is relatively more common after TBI. This is likely because of abundant diploic and dural vascularization normally present in infants and young children, notwithstanding the tight adherence of dura to the inner table of the skull.81
EDHs result from vascular injuries to dural vessels or the skull and are often associated with overlying skull fractures. They rarely occur spontaneously in patients with infections,82 sinusitis,83 vascular anomalies,84 or chronic renal failure.85–88 The classic EDH occurs beneath a temporoparietal skull fracture owing to damage to the middle meningeal artery. Separation of dura and bone is thought to occur at the time of injury rather than in a delayed fashion due to stripping of the dura from the inner table as a result of clot enlargement.89 EDHs can be classified by their radiographic progression into three appearances: type I (acute and hyperacute—day 1, associated with “swirl” of unclotted blood), type II (subacute—days 2 to 4, solid), and type III (chronic—days 7 to 20, mixed or lucent with contrast enhancement).90 They occur in 58%, 31%, and 11% of cases, respectively.
The classic clinical course of a patient with EDH was first described by Jacobson in 188691,92: initial loss of consciousness after trauma, transient complete recovery (“lucid interval”), then rapid progression of neurological deterioration. This classic presentation occurs in only 14% to 21% of patients with an EDH.93 Neurological deterioration from an expanding EDH typically results in obtundation, contralateral hemiparesis, ipsilateral oculomotor nerve paresis, decerebrate rigidity, arterial hypertension, cardiac arrhythmias, respiratory disturbances, and finally, apnea and death. Development of these symptoms depends on hematoma size94 and the presence of associated intracranial lesions.95,96 Patients with pure EDHs have an excellent prognosis after surgical evacuation, whereas those with associated intradural lesions experience good outcome in only 44% of cases.97 Although rapid diagnosis and evacuation are critical factors, data suggest that appropriate treatment (by a neurosurgeon, compared with a general surgeon in remote areas) is also important in determining patient outcome.98,99
Morbidity and mortality from a pure EDH is primarily due to delay in diagnosis and appropriate treatment. Lee and associates100 found that patients with epidural hematoma volume greater than 50 cm3 before evacuation experience worse neurological outcome and increased mortality. Delays in diagnosis of EDH are often due to so-called atypical clinical presentations. It is important for the practicing neurosurgeon to be aware, however, that the classic clinical course is far less common and that atypical presentations are far more frequent with EDH. The classic lucid interval is most common in pure EDHs that are very large and demonstrate CT signs of active bleeding (type I).94,101 However, it also occurs in patients harboring subdural hematomas93 or those with mild TBI complicated by meningitis. In fact, a review of 80 consecutive cases of EDH revealed lucid intervals in only 5 patients (6.25%).78 Instead, patients with EDH may be unconscious from the time of initial injury (23% to 44%), may regain consciousness after a brief coma (20% to 28%), or may have no loss of consciousness whatsoever (8% to 24%).77,80,102,103 Typical clinical symptoms of EDH include hemiparesis (contralateral or ipsilateral due to Kernohan’s notch),80,104 decreased level of consciousness, and dilation of the ipsilateral pupil (occurs in less that 50% of cases).101,105,106
EDH in the posterior fossa is a rare finding,107,108 accounting for about 5% of all posttraumatic intracranial mass lesions.109,110 These EDHs are particularly challenging to manage because these patients may remain conscious until late in the evolution of the hematoma, when they may suddenly lose consciousness, become apneic, and die. The neurosurgeon should be aware that these hematomas often extend into the supratentorial compartment by stripping the dura over the transverse sinus, resulting in significant intracranial hemorrhage during surgical evacuation.10 Outcome in patients with posterior fossa EDH is generally better in children and correlates with GCS on admission and CT evidence of hydrocephalus (due to compression of the fourth ventricle).111
Subdural Hematoma
Subdural hematomas (SDHs) are located between the dura and arachnoid layer and may occur as a result of arterial or venous hemorrhage. Classically, SDHs are due to tearing of bridging veins that span the subdural space to drain cortical blood directly into dural sinuses. Many SDHs, however, result from bleeding from other structures adjacent to the subdural space, such as superficial cortical vessels. SDH can be classified as acute, subacute, or chronic, although there is no uniformity of nomenclature.71,112,113 Pathologically, an acute SDH is composed of clot and blood (within 48 hours), a subacute SDH represents a mixture of clotted blood and fluid (2 to 14 days), and a chronic SDH is one that is in fluid phase (>14 days; Table 331-7).71 Clinically, an acute SDH becomes evident within 3 days of injury, subacute between 3 and 21 days, and chronic if more than 21 days pass between injury and clinical presentation. Radiographically, acute SDHs are hyperdense on CT scan, subacute are hyperdense to isodense, and chronic are hypodense relative to adjacent brain.

Acute Subdural Hematoma
Acute SDHs account for 50% to 60% of all subdural hematomas. They are most common after sudden head movements that occur with assaults or falls.79 In a review of patients with acute SDH, 72% had suffered falls or assault, whereas only 24% experienced a motor vehicle crash.33 Rarely, acute SDH may occur spontaneously (or after minor trauma) in patients receiving chronic anticoagulation therapy114,115 or after rupture of a posterior communicating artery aneurysm (only four of these cases have been seen by the senior author (JPM) in more than 30 years of practice).
Most acute SDHs result from venous vascular injury at the brain surface, resulting in two distinct pathologies.116 The first type of hematoma, produced by contact forces and associated with contusions or lacerations, results from cortical bleeding into the adjacent subdural space and is most common at the temporal pole. This complex of subdural hematoma and damaged and necrotic brain is termed burst lobe. The second type of SDH is located over the cerebral convexity and is produced by inertial forces that tear bridging veins.33,116 The underlying brain damage in this type of injury is usually milder and is primarily due to local ischemia from mass effect or compromised venous outflow. Rapid deterioration, as in the case of classic EDH, may accompany these lesions, especially if cortical arteries are ruptured. Despite the often relatively minor underlying brain damage, prognosis is generally poor in these patients unless the hematoma is rapidly evacuated.117
Cerebral ischemia plays a critical role in the pathology of SDH and has been demonstrated both experimentally118 and in postmortem studies.119 The mechanisms responsible for ischemia after SDH are poorly understood but are likely due to compressive effects of the hematoma and elevated ICP with resultant compromised CPP. Evidence of brain compression resulting in ischemia was reported by Schroder and colleagues120; in two patients with acute SDH, preoperative CBF was in the ischemic range (<18 mL/100 g per minute), and cerebral blood volume (CBV) was about half of normal. These values normalized immediately after surgical evacuation. In a small series of five patients with acute SDHs and low GCS scores (≤5), Verweij and colleagues found ICP between 40 and 80 mm Hg, CPP between 10 and 60 mm Hg, low jugular venous oxygen saturation (40% to 60%), and low cerebral blood flow (CBF) measured with laser Doppler immediately before hematoma evacuation.121 Values began to normalize with elevation of the bone flap, opening of the dura, and subsequent hematoma removal.
Timely clot evacuation (within 4 hours) generally results in significantly improved neurological outcome.122 Patients with initial CT evidence of significant hemispheric or generalized brain swelling have extremely poor outcome with or without early surgery.123 The prognosis of SDH is still poor in many cases. It is thought that the coexisting brain damage (DAI, contusion, laceration) is responsible for poor neurological function after injury. In a subset of patients, compression of the microcirculation and resultant low CBF may explain the poor clinical condition and outcome. Patients who deteriorate after a lucid interval (i.e., patients who “talk and die”) may be the subset of patients that experience this type of insult.120
Chronic Subdural Hematoma
Membrane formation around a chronic SDH is a characteristic process attributed to an inflammatory response from the dura. The inner dural layer is highly vascular, and direct contact with blood products, fibrin, and fibrin degradation products in the subdural cavity is thought to elicit a nonspecific inflammatory reaction.124 Because the arachnoid membrane has a much lower reaction potential, the inner capsule of chronic SDHs has no significant vascularity.125
Secondary enlargement of a chronic SDH is common, but the precise cause is unknown. Gardner proposed that the membrane surrounding a chronic SDH acts as an osmotic barrier, allowing cerebrospinal fluid (CSF) diffusion into the hyperosmotic hematoma.126 Zollinger and Gross also proposed an osmotic mechanism for enlargement of the hematoma; they thought that flow across the membrane was the result of an increase of osmotic pressure from a breakdown of hemoglobin molecules in red cells.127 Weir, however, could not find a significant increase in osmolality of the hematoma with increasing age, nor were there any significant differences between the osmolality of blood and hematoma.128 Sato and Suzuki used light and electron microscopy to examine the capsules of chronic subdural hematomas in 33 cases and found that in patients with neurological deficits, capillary endothelial cells in the capsule had many cytoplasmic protrusions and fenestrations, suggesting high permeability of the capillary wall.129 Observations by Yamashima and Yamamoto130 revealed that gap junctions frequently form between adjacent endothelial cells. Numerous blood components, including red blood cells and plasma, can be seen squeezing or spilling into the interstitial space of the outer membrane. It is no longer thought that an osmotic mechanism plays a significant role in hematoma enlargement. Either repeated microhemorrhages from the neocapillary network in the outer membrane or abnormally high vascular permeability is thought to be responsible for hematoma enlargement.125,131
The presentation and pathophysiology of chronic SDHs are distinct from acute SDH, and neurological signs and symptoms are typically due to extra-axial brain compression rather than neurological injury. In a study by Tanaka and coworkers,132 chronic SDHs were shown to have no significant effect on global ICP, CPP, or CBF, but instead resulted in mechanical distortion of central brain regions (e.g., thalamus) with secondary cortical effects. As a result, neurological recovery from evacuation of chronic SDHs is often excellent. Risk for recurrence of chronic SDH after surgical evacuation has been associated with hematoma width and density, lower GCS at time of presentation, ambulation within the first 72 hours, and pneumocephalus that persists beyond 7 days after surgery.133–135
Intracerebral Hematoma
Intracerebral hematomas (ICHs) account for 20% of all traumatic intracranial hematomas.136 They are associated with extensive lobar contusions, from which they are often difficult to distinguish.137,138 ICHs differ from cerebral contusions in that a large proportion of these lesions are composed of blood, but they often result from growth or coalescence of smaller cerebral contusions. By definition, an ICH is a parenchymal lesion composed of at least two-thirds blood; otherwise, the lesion is described as disrupted tissue with areas of microscopic hemorrhage.139 A hemorrhagic mass should be considered an intracerebral hematoma when there is a homogeneous collection of blood with relatively well-defined margins. Multiple intracerebral hematomas are found in about 20% of TBI cases.140
Because ICHs typically result from rupture of intrinsic cerebral vessels (a small parenchymal artery in most cases), they often arise from cerebral contusions. As a result, most traumatic ICHs occur in the orbitofrontal and temporal lobes, as do most cerebral contusions.109,136,137,141,142 Deeper ICHs, such as those occurring in the basal ganglia and internal capsule, are less common and found in about 2% of TBI patients. ICHs are most common in focal head injuries, such as missile injuries, perforating wounds, and depressed skull fractures.143 In a series of 400 civilian TBI cases with depressed skull fracture, 61% of intracranial hematomas were ICHs.144 Patients on chronic anticoagulation therapy are at increased risk for developing ICH, even after mild head injury.143 Recent studies145,146 suggest that treatment with recombinant factor VIIa may reduce expansion of traumatic intracranial hemorrhages, but randomized trials are needed to determine whether this therapy results in improved neurological outcome.
Diffuse brain injuries are the most common result of TBI and represent a clinical continuum of injury from mild concussion to persistent posttraumatic coma (see Table 331-3).147
Concussion
Concussion is the mildest form of diffuse injury and is thought to be due to rotational acceleration of the head in the absence of significant mechanical contact. In its classic form, patients with concussion experience a transient loss of consciousness followed by a rapid return to a normal state of alertness. However, concussion is not as harmless as previously thought, and repeat concussions often result in some degree of permanent neurological impairment.148–150
The pathophysiology of concussion is poorly understood and may be due to disturbances of consciousness from lesions of the brainstem and diencephalon.151 Recent studies suggest that patients with concussion often have diffuse cerebral hemisphere involvement, and brainstem lesions are far less common.152 DTI reveals signs of cytotoxic edema in the brain despite a normal head CT and GCS of 15.153,154
Diffuse Axonal Injury
DAI results from severe angular and rotational acceleration and deceleration that delivers shear and tensile forces to axons. As a result, DAI is responsible for most TBI patients that are severely impaired despite lack of gross parenchymal contusions, lacerations, or hematomas. DAI was described by Stritch in 1956155 in her report of a series of patients with severe posttraumatic dementia and “diffuse degeneration of the white matter.” Others have described this injury as shearing injury, diffuse damage to the white matter of the immediate impact type, and diffuse white matter shearing injury,1,152,156–158 but DAI is the preferred terminology. Coronal or lateral acceleration injuries produce the most severe DAIs,159 whereas acceleration in the oblique or sagittal plane results in less severe to minimal DAI.
The histologic findings of DAI have been well described and include disruption and swelling of axons, “retraction balls” (swollen proximal ends of severed axons), and punctate hemorrhages in the pons, midbrain, and corpus callosum.160 Many of these abnormalities, including axonal severing, are not present initially but develop over the course of several hours or days after injury.161 In many cases, it is difficult to distinguish axonal damage due to mechanical shearing (primary injury) from damage caused by biochemical and metabolic sequelae of TBI (secondary injury). Animal studies suggest that axonal injury is often secondary or delayed, and there is some confirmation of this in humans.161,162 These findings have critical implications for the treating physician because clinical interventions can occur before or during onset of these biochemical cascades.
DAI is often associated with punctate hemorrhages, termed Strich hemorrhages, that represent bleeding from small cerebral vessels.160 Strich hemorrhages are typically found in areas that experience maximal acceleration forces during trauma: corpus callosum, peri-third ventricular (hypothalamus, columns of the fornix, anterior commissure), internal capsule, basal ganglia, dorsolateral brainstem, and superior cerebellar peduncles. The location and severity of axonal injuries are important determinants of functional recovery. Adams and associates1 developed a grading system for patients with DAI (Table 331-8) that predicts length of coma and persistent neurological deficits.
TABLE 331-8 Neuropathologic Classification of Diffuse Axonal Injury
| GRADE | LOCALIZATION OF LESION |
|---|---|
| I | Axonal injury of parasagittal white matter of cerebral hemispheres |
| II | Grade I plus focal lesion in corpus callosum |
| III | Grade II plus focal lesion in cerebral peduncle |
From Adams JH, Doyle D, Ford I, et al. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology 1989;15:49-59.
DAI lesions are often difficult to visualize on conventional CT and are better imaged using MRI techniques. T2-weighted gradient-echo (GE) imaging is particularly sensitive for hemorrhagic lesions after DAI, whereas diffusion-weighted (DW) sequences are more effective in identifying shear injuries.163,164 DW images detect some lesions that are missed by GE sequences and correlate better with neurological prognosis following TBI.163 Recently, DTI has been used to more effectively characterize white matter lesions after TBI. DTI in the early period after TBI has been shown to correlate with GCS score and eventual outcome at discharge (using modified Rankin score).165 Xu and colleagues56 recently demonstrated that DTI is more accurate than conventional MRI in demonstrating the extent of white matter injury after severe TBI. Benson and colleagues166 found that DTI can be used to more accurately characterize injury severity, and Wang and coworkers55 found that quantitative diffusion tensor tractography in the acute period after head injury is highly predictive of outcome. As imaging techniques (and availability) continue to develop, MRI will likely play a larger role in predicting outcome and directing care for patients with DAI.
Traumatic Subarachnoid Hemorrhage and Posttraumatic Vasospasm
The centripetal theory of Ommaya and Gennarelli suggests that lesion depth is dependent on the force of injury.167 Accordingly, traumatic subarachnoid hemorrhage (SAH) results from relatively severe injury to the brain: high angular acceleration of long duration is necessary to produce a strain that causes rupture of the superficial vessels in subarachnoid cisterns. Traumatic SAH is relatively common after severe TBI, occurring in about 33% to 60% of all cases,67,168,169 and strongly correlates with worse neurological outcome.168,170–173
Posttraumatic vasospasm (PTV) is a significant secondary insult to the injured brain that is an independent predictor of permanent neurological deficit and poor outcome.170,174–181 The incidence of PTV varies by frequency of screening and diagnostic modality but is estimated to be 18.6% to 50% in the anterior circulation170,179,182–190 and 19% to 37% in the posterior circulation.180,191,192 It typically develops between 12 hours and 5 days after injury and lasts anywhere between 12 hours and 30 days.170,180,185–187,189–191,193,194 PTV can also occur in a more delayed fashion and may involve both anterior and posterior circulation arteries.195
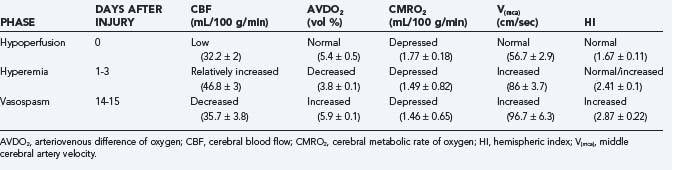
In an earlier study, Martin and colleagues used CBF and transcranial Doppler measurements to identify three different circulatory stages after severe head injury: phase I (hypoperfusion), phase II (hyperemia), and phase III (vasospasm) (Table 331-9).186 Phase I occurs on the day of injury (day 0) and is defined by low CBF, normal middle cerebral artery (MCA) velocity, normal hemispheric index (ratio of MCA velocity to internal carotid artery velocity), and normal arteriovenous difference of oxygen (AVDO2). The cerebral metabolic rate of oxygen (CMRO2) is about 50% of normal during this phase and remains depressed during the second and third phases. In phase II (relative hyperemia phase, days 1 to 3), CBF increases, AVDO2 falls, MCA velocity rises, and hemispheric index remains less than 3. In phase III (vasospasm phase, days 4 to 15), there is a fall in CBF, a further increase in MCA velocity, and a pronounced rise in the hemispheric index.
The mechanisms resulting in PTV are poorly understood but may involve extension of intradural blood into CSF spaces (subarachnoid, intraventricular, and subdural).177 Romner and associates187 found anterior circulation PTV incidence increased from 28% to 41% with traumatic SAH, a correlation that has been supported by other authors as well.177,189,196,197 Key differences do exist between traumatic and aneurysmal SAH. For example, traumatic SAH has a different distribution, often involving supratentorial, cortical convexity, sulcal and interhemispheric spaces. PTV also differs in its time course, occurring earlier and resolving more quickly on serial CT than aneurysmal SAH.198 PTV results in decreased CBF with decreased transcranial Doppler velocity. Finally, development of PTV is not always associated with significant SAH and has been reported in patients with extra-axial hematomas177,189,190,199 or no radiographic evidence of SAH whatsoever.190,199
Fukuda and colleagues reported that unlike aneurysmal SAH, in which all low-density areas on CT scans corresponded to vascular territories, low-density areas in patients with traumatic SAH were rarely associated with vascular territories.198 Instead, the low-density areas contained deep-seated or gliding contusions. As discussed in detail later, vasospasm of the large intracranial arteries is accompanied by an increase in CBV, owing to compensatory dilation of the vessels in the microcirculation. Reduced CBF in the presence of increased CBV thus supports the diagnosis of large artery spasm. Schroder and colleagues simultaneously evaluated early CBF and CBV in seven patients with severe head injury.200 These patients were selected from a larger series of 51 patients because they exhibited both nonischemic and ischemic (CBF < 18 mL/100 g per minute) areas on stable xenon CT measurements. Both CBF and CBV were significantly lower in the ischemic zones, indicating that in the early phase after injury, compromise of the microvasculature is the cause of ischemia, rather than vasospasm of the larger conductance vessels. No simultaneous studies have been performed at a later stage (i.e., when the highest incidence of vasospasm is expected).
Intraventricular Hemorrhage
About 25% of patients with severe TBI have CT evidence of intraventricular hemorrhage (IVH). Development of IVH typically requires a large force to the head and is only occasionally present after mild TBI.201 Patients with IVH are also more likely to demonstrate intraparenchymal and basal ganglia hemorrhages.202,203 Postmortem examinations have shown that most patients with primary IVH (no significant parenchymal blood) had a high incidence of damage to the septum pellucidum, choroid plexus, and subependymal vein in the fornix.204 Tearing of these veins is likely the result of negative pressure generated from a sagittal impact to the skull that transiently increases ventricular diameter.202
Although traumatic IVH has the potential of obstructing CSF flow, acute hydrocephalus is an uncommon manifestation. In a review by LeRoux and associates,203 only 4 of 43 patients with traumatic IVH required ventricular drainage for acute hydrocephalus. In a study by Hashimoto and coworkers, 20 of 32 patients with traumatic IVH did not survive their injuries.204
Normal Physiology of Cerebral Metabolism and Circulation
Cerebral Metabolism
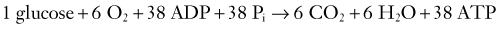
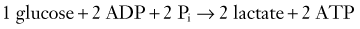
Two terms are common in reference to metabolic turnover of glucose and oxygen: the cerebral metabolic rate of oxygen (CMRO2) and the cerebral metabolic rate of glucose (CMRglc). Under normal circumstances, in awake adults, the CMRO2 is about 3.3 mg/100 g brain tissue per minute,205 and the CMRglc is 5.5 mg/100 g per minute.206 The lactate-oxygen index (LOI) is sometimes used as a measure of the ratio of the amount of glucose metabolized anaerobically to the amount metabolized aerobically (LOI = AVDL/AVDO2), although it does not accurately reflect the stoichiometry of glucose metabolism. AVDL is the arteriovenous difference of lactate, and AVDO2 is the arteriovenous difference of oxygen.206 Both are calculated by subtracting the jugular venous oxygenation (SjvO2) from the systemic arterial oxygenation (SaO2), followed by a correction for hemoglobin oxygen-carrying capacity in the latter:
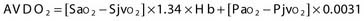
Using 2-fluoro-2-deoxy-D-glucose (FDG)–positron emission tomography (PET), Bergsneider and associates207 described a metabolic ratio (MR = CMRO2/CMRglc) to compare the amount of oxygen versus glucose utilization. Reference values of metabolism are summarized in Table 331-10. The global arteriovenous difference of glucose (AVDglc) has been estimated at 9.6 mL/dL but requires careful interpretation because reliable AVDglc values are difficult to obtain. The absolute AVDglc value is near or within the accuracy limits of plasma glucose levels of most clinical laboratories.
TABLE 331-10 Normal Values of Parameters of Cerebral Metabolism and Circulation in Healthy Adults
| PARAMETER | VALUE |
|---|---|
| CMRO2 | 3.3 ± 0.4 mL/100 g/min |
| CBF (mixed [sub]cortical flow) | 54 ± 12 mL/100 g/min |
| AVDO2 | 6.7 ± 0.8 mL/dL |
| CMRG | 5.5 ± 1.1 mg/100 g/min |
| AVDG | 9.6 ± 1.7 mL/dL |
| LOI | 0.06 ± 0.03 |
| MR | 0.49 ± 0.07 mg O2/mg glucose |
AVDG, arteriovenous difference of glucose; AVDO2, arteriovenous difference of oxygen; CBF, cerebral blood flow; CMRG, cerebral metabolic rate of glucose; CMRO2, cerebral metabolic rate of oxygen; LOI, lactate-oxygen index; MR, metabolic ratio.
Of the total energy generated, 50% is used for neurotransmitter production, release, and uptake (synaptic activity); 25% is used for maintenance and restoration of ion gradients across the cell membrane; and the remaining 25% is used for molecular transport, biosynthesis, and other unidentified processes.208,209 Although glial cells account for 50% of the brain, they have a much lower metabolic rate and account for less than 10% of total cerebral energy expenditure.209 Compared with other organs, the metabolic demand of the brain is high: the brain accounts for only 2% to 3% of total body weight and does not do any mechanical work, yet it receives 20% of cardiac output.
Regulation of Cerebral Blood Flow
Normally, CBF is regulated to provide an adequate supply of substrates to the brain. This is primarily accomplished through changes in caliber of resistance vessels, arterioles with a diameter of 30 to 300 µm.210 This process, termed autoregulation,211 occurs in response to metabolic signals (metabolic autoregulation), blood pressure (pressure autoregulation), and viscosity (viscosity autoregulation) and reacts to changes in the arterial partial pressure of CO2 (carbon dioxide reactivity).
Pressure Autoregulation
In a healthy person, pressure autoregulation is extremely effective in maintaining relatively constant CBF in the CPP range of 50 to 150 mm Hg (Fig. 331-4). When CPP drops below these limits, vessels are maximally dilated and can no longer maintain adequate CBF in the context of dropping CPP. When CPP exceeds these limits, vessels can no longer remain constricted due to high intravascular pressure and experienced forced dilation; this condition is called pressure breakthrough.
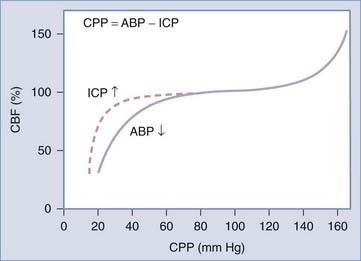
Defining autoregulation as constancy of CBF despite changes in CPP is not completely accurate however, because small changes in CBF normally occur during CPP changes within the limits of autoregulation. Therefore, some authors instead define pressure autoregulation in terms of cerebrovascular resistance (CVR), that is, the change in CVR (calculated as CPP/CBF) that occurs in response to a given change in CPP.212 Pressure autoregulation is considered intact when 0 < Δ% CPP/ Δ% CVR < 2.213–215
Viscosity Autoregulation
Poiseuille’s equation can also be used to describe the effects of viscosity on CBF. Blood viscosity changes with variations in hematocrit, gamma globulin, and plasma fibrinogen. Increased viscosity results in increased cerebral vascular resistance [CVR = 8 × 1 × viscosity/π·r4], which triggers an autoregulatory response. Under these circumstances, viscosity autoregulation responds by vasodilating and vasoconstricting in response to changes in serum viscosity to maintain relatively constant CBF.216–218
Theories of Autoregulation
Metabolic coupling is thought to be due to vasoactive metabolites released from active neurons. This concept was first introduced in 1890 by Roy and Sherrington: “The chemical products of cerebral metabolism contained in the lymph, which bathes the walls of the arterioles of the brain, can cause variations in the caliber of the cerebral vessels. In the reaction the brain possesses an intrinsic mechanism by which its vascular supply can be varied locally in correspondence with local variations of functional activity.”219 The identity of these vasoactive agents, however, remains unclear. Agents that are directly influenced by local energy metabolism, such as CO2, H+, O2, adenosine, and the ions K+ and Ca2+, have been proposed.210,220,221 However, there are problems with each of these compounds as a sole factor.209
The vascular endothelium plays an important role in maintaining the normal physiologic function of the blood vessel wall by releasing relaxing and contracting factors. Endothelial-derived relaxing factor–nitric oxide (EDRF-NO) is the primary mediator of endothelin-dependent relaxation.222 The role of EDRF-NO in autoregulation is controversial, and many authors think its main role is maintenance of basal CBF.223,224 Garthwaite and colleagues, however, found that EDRF-NO may mediate a functional coupling of metabolism and CBF in certain types of neural activation.225 During glutamate activation of the N-methyl-D-aspartate (NMDA) receptors, they noted calcium-dependent release of a substance with properties similar to that of EDRF-NO. The observed vasodilation after glutamate activation was inhibited by administration of both an NMDA antagonist and an NO synthase inhibitor, suggesting coupling of neuronal activation and CBF through EDRF-NO release.
Goadsby and colleagues simultaneously studied cerebral neuronal activity and local blood flow in cats by the induction of spreading depression (a wave of depolarization).226 Neuronal activity and CBF in the parietal cortex were measured simultaneously. They found that intravenous administration of NG-nitro-L-arginine methyl ester (1-NAME), a potent NO synthase inhibitor, resulted in a complete blockade of the hyperemia associated with spreading depression, but did not cause a change in either resting cell firing or spreading depression-evoked increases in firing rate.
Rapid elevation of transmural pressure triggers vasoconstriction (the Bayliss effect), a response that is prevented by removal of the endothelium.227,228 Pressure autoregulation is thought to be mediated by the endothelium. Two major endothelium-derived contracting factors can be identified: thromboxane A2 and endothelin.223 Martinez-Orgado and coworkers studied the influence of endothelial factors on pressure autoregulation in a porcine model.229 Middle cerebral arteries from 3- to 4-day-old piglets were cannulated, and diameter changes after transmural pressure variation were measured. Segments with endothelium showed vasodilation during pressure decrease and vasoconstriction during pressure increase, whereas segments without endothelium responded passively to pressure change. Their results suggested an endothelium-dependent autoregulatory mechanism, with involvement of NO and K-Ca channels in vasodilation during transmural pressure decrease and mediation of vasoconstriction by endothelin-1 (through endothelin A receptors) and prostanoids during pressure increase.
Winn and colleagues have extensively studied the role of adenosine in regulating pial vessel diameter and vascular resistance and have implicated its role as a metabolic trigger for autoregulation.230–234 The mechanisms of its actions are not completely understood but may include nitric oxide release from astrocytes,235 recruitment of cyclic guanosine monophosphate–dependent pathways,236 changes in blood-brain barrier permeability to excitatory amino acids,237 and direct activity on arteriolar adenosine receptors.232
Carbon Dioxide Reactivity
The effects of hypocarbia and hypercarbia are mediated by pH changes in the perivascular space. The effects of CO2 changes on blood vessel diameter persist for less than 20 to 24 hours; at a constant level of PacO2, the pH in the perivascular space normalizes, and cerebral blood vessel diameter returns to baseline.238 To maintain a constant supply of substrates to support metabolism (CMRO2), changes in CBF due to hypocarbia or hypercarbia are compensated for by changes in AVDO2. Again, this is fundamentally different from metabolic, pressure, and viscosity autoregulation, whereby AVDO2 remains constant and CBF is “tuned” to metabolism.
Cerebral Blood Flow and Cerebral Blood Volume
Alterations in vascular caliber not only affect brain perfusion but also change CBV. Changes in CBV can have dramatic effects on ICP, which may be critical when managing patients with severe TBI. CBV is determined by CBF, and the mean transit time of blood through the cerebral vasculature (MTT): CBV = CBF × MTT. Most of the intracranial blood volume is present in vessels with diameters between 30 and 300 µm. Changes in vessel caliber primarily occur in arterioles (200 µm), whereas the diameter of venules remains more or less constant.210
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


