4
Confusional States
A confusional state, sometimes referred to as encephalopathy or delirium, is a state in which the level of consciousness is depressed, but to a lesser extent than in coma (unarousable unresponsiveness; see Chapter 3). In confusional states, responses to stimulation are at least semi-purposeful, whereas in coma, patients fail to respond to even painful stimulation or do so only in reflex fashion. Thus the difference between a confusional state and coma is largely one of degree, and the causes overlap extensively.
APPROACH TO DIAGNOSIS
Evaluation of a patient with altered consciousness is aimed first at characterizing the nature of the disorder (confusional state, coma, or a more chronic condition, such as dementia) and second at determining the cause. If the patient is the only source of information, little history may be available from him or her. However, old medical charts and the patient’s clothing and other personal effects can provide diagnostic clues, and the general physical examination may be similarly helpful.
A confusional state can be most readily distinguished from dementia by the time course of the impairment: confusional states are acute or subacute in onset, typically developing over hours to days, whereas dementia is a chronic disorder that evolves over months or years.
Certain causes of confusional state must be identified urgently because they may lead rapidly to severe structural brain damage or death, and prompt treatment can prevent these complications: examples include hypoglycemia, bacterial meningitis, subarachnoid hemorrhage, traumatic intracranial hemorrhage, and Wernicke encephalopathy (Table 4-1).
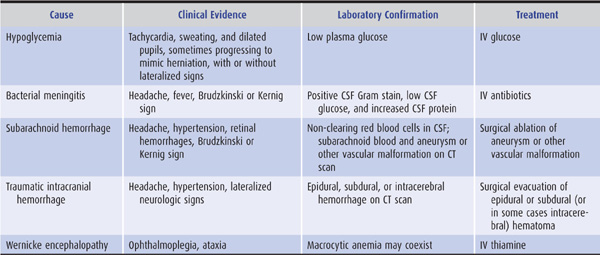
Table 4-1. Most urgent causes of confusional states.
HISTORY
History of Present Illness
The history should establish the time course of the disorder and provide clues to its nature and cause. Confusional states are acute to subacute in onset, whereas dementias are chronic disorders. In either case, the observations of others may be the only history available. It is, therefore, useful to have access to a relative or friend who can furnish details about the patient’s previous level of functioning, the time when dysfunction became evident, and the nature of observed changes.
Past History
Preexisting conditions that predispose to confusional states should be noted. Examples include alcoholism (intoxication or withdrawal, or Wernicke encephalopathy), other drug abuse (intoxication or infection), diabetes (hypo- or hyperglycemia), heart disease (stroke), epilepsy (seizures or postictal state), and head trauma (concussion, intracranial hemorrhage). A thorough medication history is also important, because numerous therapeutic drugs can impair consciousness as a side effect.
GENERAL PHYSICAL EXAMINATION
Findings on general physical examination of a confused patient that suggest specific causes are listed in Table 4-2.

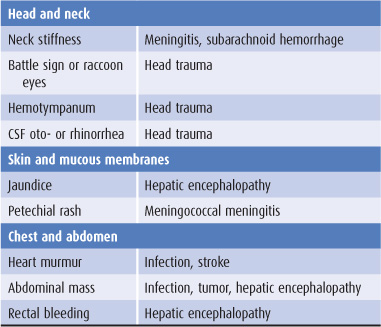
Table 4-2. General physical examination in confusional states.
NEUROLOGIC EXAMINATION
Mental Status Examination
The mental status exam of a patient with a possible confusional state should focus on confirming whether the level of consciousness is indeed depressed by evaluating the following functions.
A. Wakefulness—In confusional states, the patient often appears sleepy; this may alternate with apparent hyper-alertness, which is, however, belied by defects in orientation and attention (discussed later).
B. Arousability—In mild confusional states, the patient may still be readily arousable when spoken to or lightly shaken. As consciousness is further impaired, the intensity of stimulation required for arousal increases, the duration of arousal declines, and the responses elicited become less purposeful.
C. Orientation—With a confusional state, the patient loses orientation to time and later to place.
D. Attention—Confused patients are inattentive, as demonstrated by the inability to immediately repeat a list of numbers or words.
E. Memory—Confusion impairs short-term memory so that the patient cannot remember a short list of items when asked to repeat them after a delay of several minutes.
One pitfall to avoid in the assessment of seemingly confused patients is to mistake receptive or fluent (Wernicke) aphasia for confusion. Although patients with this aphasia cannot comprehend written or spoken language and may speak incomprehensibly, they appear normally awake and alert, can respond appropriately to nonverbal commands (such as gestures), and usually have associated right-sided neurologic abnormalities such as hemiparesis, hemisensory deficit, and visual field deficits.
Signs of Diffuse Versus Focal Disorders Causing Confusional States
Certain findings on neurologic examination help to distinguish diffuse (including metabolic) from focal (including mass) lesions as the likely cause of a confusional state.
A. Diffuse disorders—Findings suggesting a diffuse disorder include fever or hypothermia, nystagmus, tremor, asterixis, and myoclonus.
B. Focal disorders—Findings suggesting a focal disorder include signs of head trauma, papilledema, hemiparesis, focal seizures, asymmetric hyperreflexia, and unilateral Babinski signs.
Signs of Specific Disorders Causing Confusional States
Findings on neurologic examination of a confused patient that suggest specific causes of a confusional state are listed in Table 4-3.
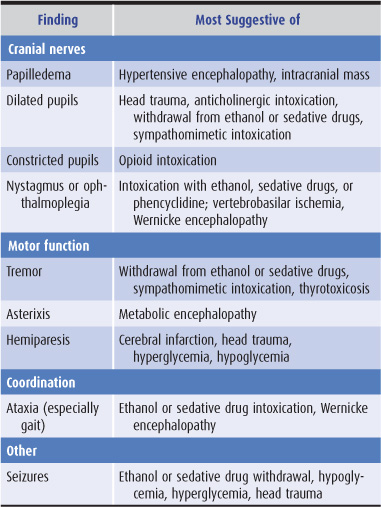
Table 4-3. Neurologic examination in confusional states.
LABORATORY INVESTIGATIONS
Laboratory tests that suggest specific causes of a confusional state are listed in Table 4-4. Cerebrospinal fluid (CSF) profiles in disorders associated with confusional states are described in more detail in Table 4-5.
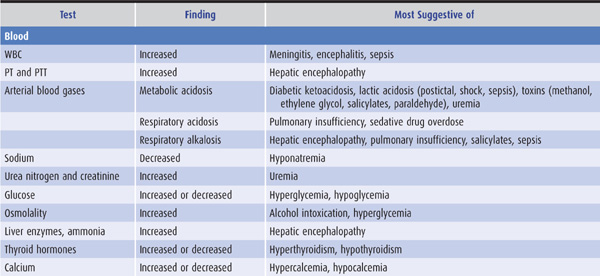
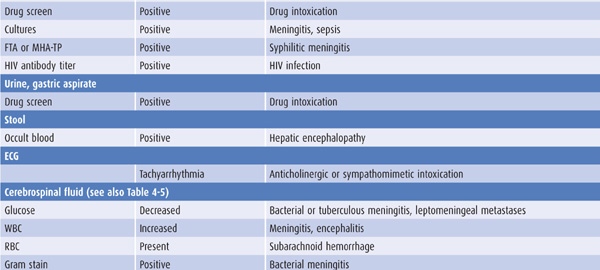
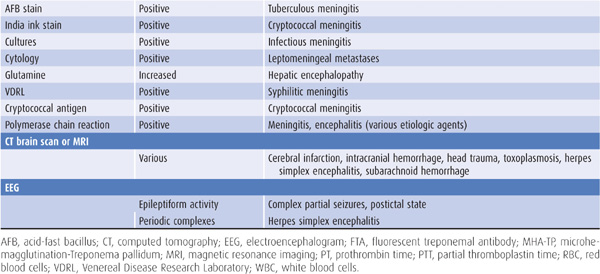
Table 4-4. Laboratory studies in confusional states.
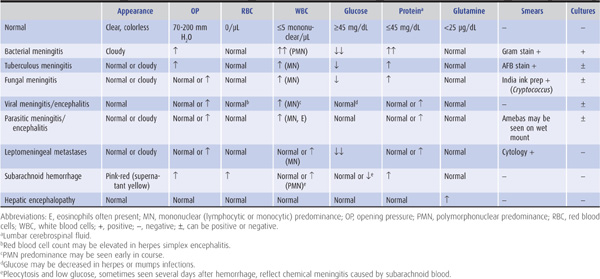
Table 4-5. Cerebrospinal fluid (CSF) profiles in confusional states.
CAUSES OF CONFUSIONAL STATES
DRUGS
Many drugs can cause confusional states, especially when taken in greater than customary doses, in combination with other drugs, by patients with altered drug metabolism from hepatic or renal failure, by the elderly, or in the setting of preexisting cognitive impairment. Evaluation of any patient with a confusional state should always include a thorough review of medications, including both prescribed and over-the-counter preparations. For a variety of reasons, “recreational” and psychotherapeutic drugs are the most likely to produce altered consciousness and are therefore emphasized here.
ALCOHOL INTOXICATION
Ethyl alcohol (ethanol) intoxication produces a confusional state with nystagmus, dysarthria, and limb and gait ataxia. In nonalcoholics, signs correlate roughly with blood alcohol levels, but chronic alcoholics, who have developed tolerance, may have very high levels without appearing intoxicated. Laboratory studies useful in confirming the diagnosis include blood alcohol levels and serum osmolality. In alcohol intoxication, serum osmolality determined by direct measurement exceeds the calculated osmolality (2 × serum sodium + 1/20 serum glucose + 1/3 serum urea nitrogen) by 22 mosm/L for every 100 mg/dL of alcohol present. Intoxicated patients are at high risk for head trauma. Alcohol ingestion may cause life-threatening hypoglycemia, and chronic alcoholism increases the risk of bacterial meningitis. Treatment is not required unless a withdrawal syndrome ensues, but alcoholic patients should receive thiamine to prevent malnutrition-related Wernicke encephalopathy (see next section).
ALCOHOL WITHDRAWAL
Three common withdrawal syndromes are recognized (Figure 4-1). Patients with these syndromes are also at risk for Wernicke encephalopathy and should be given thia-mine (typically 100 mg/d, by the intravenous or intramuscular route, until a normal diet is restored).
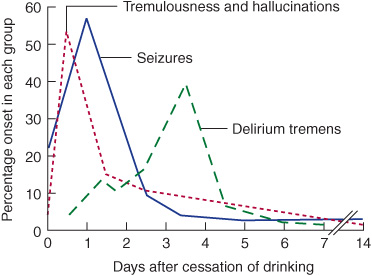
Figure 4-1. Alcohol withdrawal syndromes in relation to the time since cessation of drinking. (Data from Victor M, Adams RD. The effect of alcohol on the nervous system. Res Publ Assoc Res Nerv Ment Dis. 1952;32:526-573.)
Tremulousness & Hallucinations
This self-limited condition occurs within 2 days after cessation of drinking and is characterized by tremulousness, agitation, anorexia, nausea, insomnia, tachycardia, and hypertension. Confusion, if present, is mild. Illusions and hallucinations, usually visual, occur in approximately 25% of patients. Lorazepam 1 to 4 mg or diazepam 5 to 20 mg given intravenously every 5 to 15 minutes until calm and hourly thereafter to maintain light sedation will terminate the syndrome and prevent more serious consequences of withdrawal.
Seizures
Alcohol withdrawal seizures occur within 48 hours of abstinence and within 7 to 24 hours in approximately two-thirds of cases. Roughly 40% of patients who experience seizures have a single seizure; more than 90% have between one and six seizures. In approximately 85% of cases, the interval between the first and last seizures is 6 hours or less. Treatment is not usually required, as seizures cease spontaneously in most cases, but lorazepam 2 mg intravenously may reduce the number of seizures that occur. Unusual features such as focal seizures, prolonged duration of seizures (>6 to 12 hours), more than six seizures, status epilepticus, or a prolonged postictal state should prompt a search for other causes or complicating factors, such as head trauma or infection. The patient should be observed for 6 to 12 hours after the onset of seizures to make certain that atypical features suggesting another cause do not develop.
Delirium Tremens
This most serious ethanol withdrawal syndrome typically begins 3 to 5 days after cessation of drinking and lasts for up to 72 hours. It is characterized by confusion, agitation, fever, sweating, tachycardia, hypertension, and hallucinations. Death may result from concomitant infection, pancreatitis, cardiovascular collapse, or trauma. Treatment consists of lorazepam or diazepam as described previously for tremulousness and hallucinations and correction of fluid and electrolyte abnormalities and hypoglycemia, if present. Concomitant β-adrenergic receptor blockade with atenolol 50 to 100 mg/d may be useful for patients with persistent hypertension or tachycardia.
SEDATIVE DRUG INTOXICATION
Sedative drugs include barbiturates, benzodiazepines, gamma hydroxybutyrate (GHB), propofol (Diprivan), methaqualone, glutethimide, and chloral hydrate. The classic signs of sedative drug intoxication are confusional state or coma, respiratory depression, hypotension, hypothermia, reactive pupils, nystagmus or absence of ocular movements, ataxia, dysarthria, and hyporeflexia. Glutethimide or very high doses of barbiturates may produce large, fixed pupils, and decerebrate or decorticate posturing can occur in sedative drug-induced coma. The diagnosis of sedative drug intoxication can be confirmed by toxicologic analysis of blood, urine, or gastric aspirate, but blood levels of short-acting sedatives do not correlate with clinical severity.
Management is directed at supporting the patient’s respiratory and circulatory function while the drug is being cleared, primarily by hepatic metabolism. In addition, patients with benzodiazepine intoxication can be treated with the benzodiazepine receptor antagonist flumazenil 1 to 5 mg intravenously over 2 to 10 minutes, repeated at 20- to 30-minute intervals as needed.
Complications of sedative drug intoxication include aspiration pneumonia, hypotension, and renal failure. However, barring the development of such complications, patients who arrive at the hospital with adequate cardio-pulmonary function should survive without sequelae.
SEDATIVE DRUG WITHDRAWAL
Like alcohol, sedative drugs can produce withdrawal syndromes manifested by confusional states, seizures, or delirium tremens when intake is stopped abruptly. The likelihood and severity of withdrawal syndromes depend on the duration of drug intake and the dose and half-life of the drug and are greatest in patients taking large doses of intermediate- or short-acting drugs for at least several weeks. Withdrawal syndromes typically develop 1 to 3 days after cessation of short-acting sedatives but may not appear until 1 week or more with longer-acting drugs. Sedative drug withdrawal can be confirmed by the failure of a normally sedating or hypnotic dose to produce signs of sedative drug intoxication (sedation, nystagmus, dysarthria, or ataxia). Symptoms and signs of withdrawal are usually self-limited, but myoclonus and seizures—most common in patients taking several times a drug’s sedative dose daily—may require treatment.
OPIATES
Opiates (narcotics) include morphine, heroin, codeine, hydromorphone (Dilaudid), oxycodone (OxyContin), hydrocodone (Vicodin), meperidine (Demerol), fentanyl, and methadone. These drugs can produce analgesia, mood changes, confusional states, coma, respiratory depression, pulmonary edema, nausea and vomiting, pupillary constriction, hypotension, urinary retention, and reduced gastrointestinal motility. Their chronic use is associated with tolerance and physical dependence.
Treatment consists of intravenous administration of naloxone 0.4 to 0.8 mg and sometimes ventilatory support. Because the action of naloxone may be as short as 1 hour—and many opiates are longer-acting—naloxone should be readministered as the patient’s condition dictates. With appropriate treatment, patients should recover uneventfully.
ANTICHOLINERGICS
Muscarinic anticholinergic drugs are used to treat parkinsonism (eg, trihexyphenidyl, or Artane), motion sickness (eg, dimenhydrinate, or Dramamine), allergies (eg, diphenhydramine, or Benadryl), and gastrointestinal disturbances (eg, dicyclomine, or Bentyl). Antipsychotic drugs, tricyclic antidepressants, and many antihistamines also have prominent anticholinergic activity. Overdose with any of these agents can produce a confusional state with agitation, hallucinations, fixed and dilated pupils, blurred vision, dry skin and mucous membranes, flushing, fever, urinary retention, and tachycardia. In some cases, the diagnosis can be confirmed by toxicologic analysis of blood or urine. Symptoms usually resolve spontaneously, but treatment may be required, especially if life-threatening cardiac arrhythmias occur. In such cases, the cholinesterase inhibitor physostigmine can reverse the abnormality by interfering with the breakdown of acetylcholine. However, physostigmine may produce bradycardia and seizures, so it is rarely used.
SYMPATHOMIMETICS
Sympathomimetics include cocaine, amphetamine, methamphetamine, 3,4-methylenedioxymethamphetmaine (ecstasy), dextroamphetamine (Dexedrine), methylphenidate (Ritalin), phentermine, fenfluramine, ephedrine, and antidepressants. Some tricyclic antidepressants also have anticholinergic effects. Sympathomimetic intoxication can produce a confusional state with hallucinations, motor hyperactivity, stereotypic behavior, and paranoid psychosis. Physical examination typically shows tachycardia, hypertension, and dilated pupils. Hyperthermia, tremor, seizures, and cardiac arrhythmias may occur. In addition, cocaine or amphetamine use can be associated with stroke.
Agitation may be treated with benzodiazepines, psychosis with haloperidol, and hypertension with sodium nitroprusside or phentolamine.
HALLUCINOGENS
Hallucinogens include lysergide acid diethylamide (LSD), psilocybin, mescaline, phencyclidine (PCP), ketamine, ibogaine, and bufotenin. Most do not produce confusional states that come to medical attention, but PCP can be an exception. Clinical features of PCP intoxication include drowsiness, agitation, disorientation, amnesia, hallucinations, paranoia, and violent behavior. Neurologic examination may show large or small pupils, horizontal and vertical nystagmus, ataxia, increased muscle tone, analgesia, hyper-reflexia, and myoclonus. In severe cases, complications include hypertension, malignant hyperthermia, status epilepticus, coma, and death. Benzodiazepines may be useful for sedation and for treating muscle spasms, and antihypertensives, anticonvulsants, and dantrolene (for malignant hyperthermia) may be required. Symptoms and signs usually resolve within 24 hours.
INHALANTS
These include volatile solvents (eg, glue), volatile nitrites (eg, amyl nitrite), anesthetics (eg, ether, chloroform, nitrous oxide), and propellants. Their pharmacologic actions are diverse, but most can produce euphoria followed by depression, and sometimes respiratory compromise. Withdrawal may be associated with irritability, anxiety, tremor, and seizures. There is no specific treatment.
ENDOCRINE DISORDERS
HYPOTHYROIDISM
The most common cause of hypothyroidism is Hashimoto thyroiditis, an autoimmune disorder. Symptoms and signs include fatigue, depression, weight gain, constipation, bradycardia, dry skin, and hair loss (Figure 4-2). Profound hypothyroidism may produce a confusional state, coma, or dementia. Cognitive disturbances include flat affect, psychomotor retardation, agitation, and psychosis (myxedema madness). The neurologic examination may show dysarthria, deafness, or ataxia, but the most characteristic abnormality is delayed relaxation of the tendon reflexes. Untreated, the condition can progress to seizures, coma, and death.
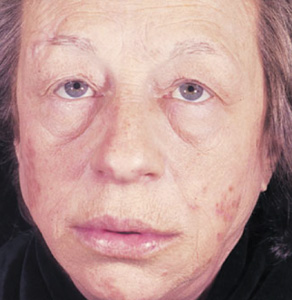
Figure 4-2. Clinical features of hypothyroidism. The patient shows a lack of facial expression, together with pallor, dry skin, loss of hair in the lateral eyebrows, facial puffiness, broadening of the nose, and drooping eyelids. (Wolff K, Goldsmith LA, Katz, et al. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York: McGraw-Hill, 2007.)
Laboratory abnormalities include low serum thyroid hormone—triiodothyronine (T3) and tetraiodothyronine (T4)—levels and elevated thyroid-stimulating hormone (TSH). Hypothermia, hypoglycemia, hyponatremia, and respiratory acidosis may occur. CSF protein is typically elevated, and CSF pressure is occasionally increased. Treatment is of the precipitating cause and the underlying thyroid disorder. In severe myxedema madness or coma, this involves intravenous administration of levothyroxine (400 μg, then 50-100 μg daily) and hydrocortisone (100 mg, then 25-50 mg every 8 hours) for associated adrenal insufficiency.
HYPERTHYROIDISM
Hyperthyroidism is most often due to Graves disease, an autoimmune disorder, which causes anxiety, palpitations, sweating, and weight loss. Acute exacerbation of hyperthyroidism (Figure 4-3) may cause a confusional state, coma, or death. In younger patients, agitation, hallucinations, and psychosis are common (activated thyrotoxic crisis), whereas those older than age 50 tend to be apathetic and depressed (apathetic thyrotoxic crisis). Seizures may occur. Neurologic examination shows an exaggerated physiologic (action) tremor and hyperreflexia, but ankle clonus and extensor plantar responses are rare. The diagnosis is confirmed by elevated serum T4, free T4, T3 and free T3, and low serum TSH. Treatment includes correction of hyperthermia, fluid and electrolyte disorders, cardiac arrhythmias, and congestive heart failure, and administration of antithyroid drugs (propylthiouracil or methimazole) and iodide to inhibit thyroid hormone synthesis and secretion, cholestyramine to promote T4 elimination, propranolol for tachycardia, and hydrocortisone for associated adrenal insufficiency. The underlying disorder that precipitated thyrotoxic crisis should also be sought and corrected.
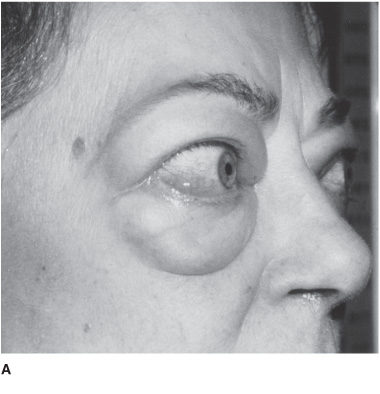
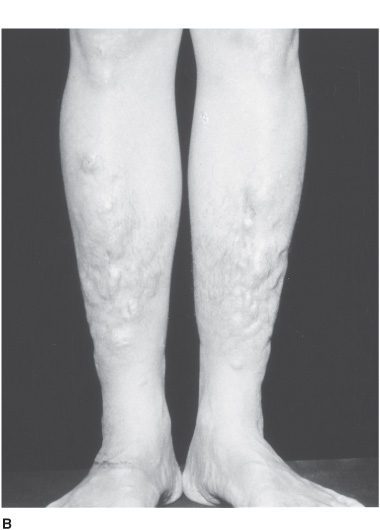
Figure 4-3. Clinical features of hyperthyroidism. The patient shows (A) ophthalmopathy with exophthalmos (proptosis), and (B) pretibial myxedema. (From Brunicardi CF, Andersen DK, Billiar TR, et al. Schwartz’s Principles of Surgery. 9th ed. New York: McGraw Hill, 2009.)
HYPOGLYCEMIA
Prompt treatment of hypoglycemia is essential because hypoglycemic encephalopathy may progress rapidly from a reversible to an irreversible stage, and definitive therapy can be quickly and easily administered.
The most common cause is insulin overdose in diabetic patients, but oral hypoglycemic drugs, alcoholism, malnutrition, hepatic failure, insulinoma, and non–insulin-secreting fibromas, sarcomas, or fibrosarcomas may also be responsible. Neurologic symptoms develop over minutes to hours. Although no strict correlation between blood glucose levels and the severity of neurologic dysfunction can be demonstrated, prolonged hypoglycemia at levels of 30 mg/dL or lower invariably leads to irreversible brain damage.
Clinical Findings
Early signs of hypoglycemia include tachycardia, sweating, and pupillary dilation, which may be followed by a confusional state with somnolence or agitation (Figure 4-4). Neurologic dysfunction progresses in a rostral-caudal fashion (see Chapter 3, Coma) and may mimic a mass lesion causing transtentorial herniation. Coma ensues with spasticity, extensor plantar responses, and decorticate or decerebrate posturing. Signs of brainstem dysfunction subsequently appear, including abnormal ocular movements and loss of pupillary reflexes. Respiratory depression, bradycardia, hypotonia, and hyporeflexia ultimately supervene, at which point irreversible brain damage is imminent. Hypoglycemic coma may be associated with focal neurologic signs and focal or generalized seizures.
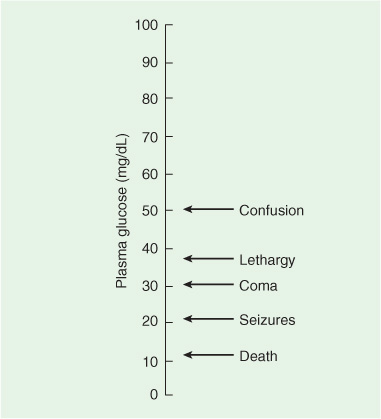
Figure 4-4. Relationship between blood glucose concentrations and impaired consciousness. Note that once hypoglycemia becomes symptomatic, even slight further decreases in plasma glucose lead to increasingly severe neurologic complications. (Modified from Barrett KE, Barman SM, Boitano S, Brooks H. Ganong’s Review of Medical Physiology. 23rd ed. New York: McGraw-Hill, 2009.)
Treatment
The diagnosis is confirmed by measuring blood glucose levels, but intravenous glucose (50 mL of 50% dextrose) should be given immediately, without waiting for the blood glucose level to be measured. Improvement in the level of consciousness is evident within minutes after glucose administration in patients with reversible hypoglycemic encephalopathy. The consequences of inadvertently worsening what later proves to be hyperglycemic encephalopathy are never as serious as those of failure to treat hypoglycemia.
HYPERGLYCEMIA
Two hyperglycemic syndromes, diabetic ketoacidosis and hyperosmolar nonketotic hyperglycemia, can produce encephalopathy or coma. Either syndrome, distinguished by a variety of clinical and laboratory features (Table 4-6), may be the presenting manifestation of diabetes. Impaired cerebral metabolism, intravascular coagulation from hyperviscosity, and brain edema from rapid correction of hyperglycemia contribute to pathogenesis. Whereas the severity of hyperosmolarity correlates well with depression of consciousness, the degree of systemic acidosis does not.
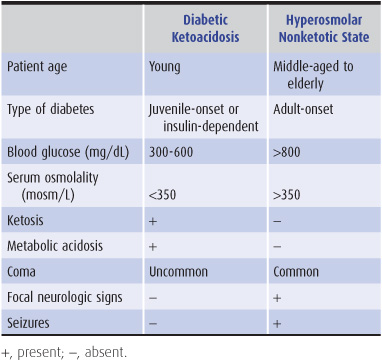
Table 4-6. Features of hyperglycemic encephalopathies.
Clinical Findings
Symptoms include blurred vision, dry skin, anorexia, polyuria, and polydipsia. Physical examination may show hypotension and other signs of dehydration, especially in hyperosmolar nonketotic hyperglycemia. Deep, rapid (Kussmaul) respiration characterizes diabetic ketoacidosis. Impairment of consciousness varies from mild confusion to coma. Focal neurologic signs and generalized or focal seizures are common in hyperosmolar nonketotic hyperglycemia. Laboratory findings are summarized in Table 4-6.
Treatment
Treatment of diabetic ketoacidosis includes intravenous regular insulin (0.15 units/kg, then 0.1 units/kg/h), fluid (0.9% saline, 1 L/h for 1-2 hours, then 300-400 mL/h), potassium (10-30 mEq/h for 2 hours, starting after acidosis has begun to resolve), and antibiotics for concomitant infections. Treatment is adjusted as needed, based on monitoring of urine glucose and ketones, arterial pH, and blood glucose, acetone, bicarbonate, electrolytes, and urea nitrogen. Deaths are usually related to sepsis, cardiovascular or cerebrovascular complications, or renal failure. In hyperosmolar nonketotic hyperglycemia, fluid replacement is most important; 0.5 N saline is administered, except to patients with circulatory collapse, who should receive normal saline. Potassium and, if indicated, phosphate should be replaced. Less insulin is required than for diabetic ketoacidosis. When death occurs, it is usually caused by coexisting disease or delayed treatment due to misdiagnosis.
HYPOADRENALISM
Adrenocortical insufficiency (Addison disease) produces fatigue, weakness, weight loss, anorexia, hyperpigmentation of the skin, hypotension, nausea and vomiting, abdominal pain, and diarrhea or constipation. Neurologic manifestations include confusional states, seizures, or coma. Treatment is administration of hydrocortisone and correction of hypovolemia, hypoglycemia, electrolyte disturbances, and precipitating illnesses.
HYPERADRENALISM
Hyperadrenalism (Cushing syndrome) usually results from the administration of exogenous glucocorticoids. Clinical features include moon facies with facial flushing (Figure 4-5), truncal obesity, hirsutism, menstrual irregularities, hypertension, weakness, cutaneous striae, acne, and ecchymoses. Neuropsychiatric disturbances are common and include depression or euphoria, anxiety, irritability, memory impairment, psychosis, delusions, and hallucinations. The diagnosis can be confirmed by a dexamethasone suppression test, 24-hour urine free cortisol level, or late night salivary cortisol assay. Measurement of serum adrenocorticotropic hormone (ACTH) distinguishes adrenal from pituitary causes of hyperadrenalism, and magnetic resonance imaging (MRI) is used to localize pituitary or other ACTH-secreting tumors. Treatment options depend on the cause and include transphenoidal resection or stereotactic radiotherapy of pituitary adenomas and laparoscopic resection of cortisol-secreting adrenal neoplasms or ectopic ACTH-secreting tumors.
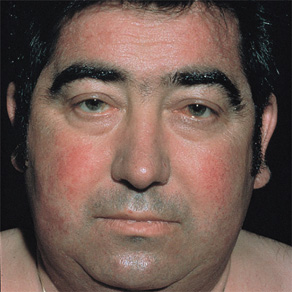
Figure 4-5. Moon (round, full, puffy) facies and facial flushing in Cushing syndrome. (From Wolff K, Johnson RA. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 6th ed. New York: McGraw-Hill; 2009.)
ELECTROLYTE DISORDERS
HYPONATREMIA
Clinical Findings
Hyponatremia, particularly when acute, produces brain swelling because of hypoosmolality of the extracellular fluid. Causes include hypothyroidism, adrenal insufficiency, drugs (eg, thiazide diuretics, nonsteriodal anti-inflammatory drugs, ecstasy), and the syndrome of inappropriate antidiuretic hormone (ADH) secretion (SIADH). Hyponatremia produces headache, lethargy, confusion, weakness, muscle cramps, nausea, and vomiting. Neurologic signs include confusional state or coma, papilledema, tremor, asterixis, rigidity, extensor plantar responses, focal or generalized seizures, and occasionally focal neurologic deficits. Neurologic complications are usually associated with serum sodium levels less than 120 mEq/L (Figure 4-6), but may be seen after a rapid fall to 130 mEq/L; conversely, chronic hyponatremia with levels as low as 110 mEq/L may be asymptomatic.
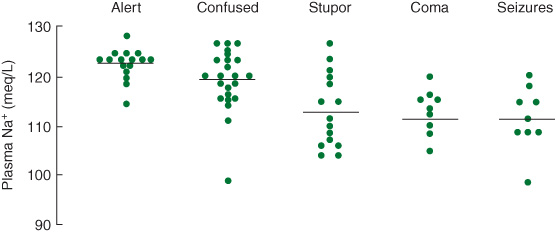
Figure 4-6. Relationship between serum sodium concentration and neurologic manifestations of hyponatremia. (Reproduced, with permission, from Arieff AI, Llach F, Massry SG. Neurologic manifestations and morbidity of hyponatremia: correlation with brain water and electrolytes. Medicine. 1976;55:121-129.)
Treatment
Treatment is most effective when the underlying cause of hyponatremia is corrected. Immediate management includes water restriction or, for severe symptoms, infusion of hypertonic saline with or without intravenous furosemide. Excessively rapid correction of hyponatremia may lead to central pontine myelinolysis (osmotic demyelination syndrome), a disorder of white matter characterized by a confusional state, paraparesis or quad-riparesis, dysarthria, dysphagia, hyper- or hyporeflexia, and extensor plantar responses. Severe cases can result in the locked-in syndrome (see Chapter 3, Coma), coma, or death. MRI may show pontine and extrapontine white matter lesions. There is no treatment for central pontine myelinolysis, so prevention is essential and may best be achieved by restricting water intake and using small amounts of hypertonic saline to raise the serum sodium concentration to between 125 and 130 mmol/L, at a rate not exceeding 8 mmol/L/d.
HYPERCALCEMIA
Hypercalcemia may result from primary hyperparathyroidism, multiple myeloma, or tumors that secrete parathyroid hormone-related protein. Symptoms include thirst, polyuria, constipation, nausea and vomiting, abdominal pain, anorexia, and flank pain from nephrolithiasis. Neurologic symptoms are always present with serum calcium levels higher than 17 mg/dL (8.5 mEq/L) and include headache, weakness, and lethargy.
Physical examination may show dehydration, abdominal distention, focal neurologic signs, myopathic weakness, and a confusional state that can progress to coma. Seizures are rare. The myopathy spares bulbar muscles, and tendon reflexes are usually normal. The diagnosis is confirmed by an elevated serum calcium level, and sometimes increased parathyroid hormone levels and a shortened QT interval on the electrocardiogram (ECG). Severe hypercalcemia in patients with normal cardiac and renal function is treated by vigorous intravenous hydration with 0.45% or 0.9% saline and usually requires central venous pressure monitoring. Bisphosphonates are added to treat hypercalcemia associated with malignancy.
HYPOCALCEMIA
Symptoms include irritability, delirium, psychosis with hallucinations, depression, nausea and vomiting, abdominal pain, and paresthesias of the circumoral region and distal extremities. The most characteristic physical signs are those of overt or latent tetany. Neural hyperexcitability is exhibited by contraction of facial muscles in response to percussion of the facial (VII) nerve anterior to the ear (Chvostek sign). Carpopedal spasm (Figure 4-7) may occur spontaneously or after tourniquet-induced limb ischemia (Trousseau sign). Cataracts and papilledema are sometimes present, and chorea is reported. Seizures or laryngospasm can be life-threatening. Serum calcium levels are below 9 mg/dL (4.5 mEq/L), but total serum calcium is also decreased in the setting of hypoalbuminemia without affecting ionized calcium, and hypocalcemia in this case is asymptomatic. The ECG may show a prolonged QT interval. Treatment is with intravenous calcium gluconate 10 to 15 mg/kg intravenously infused over 4 to 6 hours. Seizures, if present, are treated with phenytoin or phenobarbital.
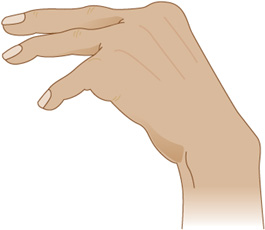
Figure 4-7. Carpal spasm, a sign of tetany (neuronal hyperexcitability) in hypocalcemia. (From Gardner DG, Shoback D. Greenspan’s Basic & Clinical Endocrinology, 8th edition. New York: McGraw-Hill, 2007.)
NUTRITIONAL DISORDERS
WERNICKE ENCEPHALOPATHY
Wernicke encephalopathy is usually a complication of chronic alcoholism, but also occurs in other disorders associated with malnutrition, such as cancer, and after bariatric surgery. It is caused by deficiency of thiamine (vitamin B1). Pathologic features include neuronal loss, demyelination, and gliosis in periventricular gray matter. Proliferation of small blood vessels and petechial hemorrhages may be seen. The areas most commonly involved are the medial thalamus, mammillary bodies, periaqueductal gray matter, cerebellar vermis, and oculomotor, abducens, and vestibular nuclei.
Clinical Findings
The classic syndrome comprises the triad of ophthalmoplegia, ataxia, and confusional state. The most common ocular abnormalities are nystagmus, abducens (VI) nerve palsy, and horizontal or combined horizontal–vertical gaze palsy. Ataxia affects gait primarily; ataxia of the arms is uncommon, as is dysarthria. The mental status examination reveals global confusion with a prominent disorder of immediate recall and recent memory. The confusional state progresses to coma in a small percentage of patients. Most patients have associated neuropathy with absent ankle jerks. Hypothermia and hypotension may occur because of hypothalamic involvement. Pupillary abnormalities, including mild anisocoria, or a sluggish reaction to light, are occasionally seen. The peripheral blood smear may show macrocytic anemia, and MRI may show atrophy of the mammillary bodies (Figure 4-8).
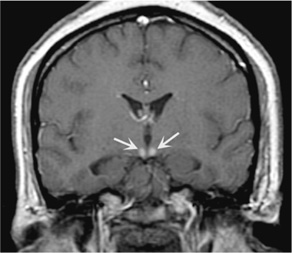
Figure 4-8. Coronal T1-weighted MRI with contrast showing abnormal enhancement of the mammillary bodies (arrows) in a patient with Wernicke encephalopathy. (From Fauci A, Braunwald E, Kasper D, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York: McGraw-Hill, 2008.)
Treatment
Treatment requires prompt administration of thiamine. An initial dose of 100 mg is given intravenously, before or with dextrose, to avoid precipitating or exacerbating the disorder. Parenteral thiamine is continued for several days. The maintenance requirement for thiamine, approximately 1 mg/d, is usually available in the diet, although enteric absorption of thiamine is impaired in alcoholics.
After treatment, ocular abnormalities usually begin to improve within 1 day and ataxia and confusion within 1 week. Ophthalmoplegia, vertical nystagmus, and acute confusion are entirely reversible, usually within 1 month. Horizontal nystagmus and ataxia, however, resolve completely in only approximately 40% of cases. The major long-term complication of Wernicke encephalopathy is Korsakoff syndrome (see Chapter 5, Dementia & Amnestic Disorders).
VITAMIN B12 DEFICIENCY
Vitamin B12 (cyanocobalamin) deficiency produces peripheral neuropathy, subacute combined degeneration of the spinal cord (combined systems disease) affecting the corticospinal tracts and dorsal columns, nutritional amblyopia (visual loss), and cognitive dysfunction that ranges from a mild confusional state to dementia or psychosis (megaloblastic madness). Neurologic abnormalities may precede the development of macrocytic anemia. The most frequent cause of vitamin B12 deficiency is pernicious anemia, a defect in the production of intrinsic factor associated with atrophic gastritis, anti-parietal cell antibodies, and achlorhydria, which is most common in those of northern European ancestry. Other causes include gastric resection and vegan diet.
Clinical Findings
The presenting symptoms are usually caused by anemia or orthostatic lightheadedness, but may also be neurologic. Distal paresthesias, gait ataxia, a bandlike sensation of tightness around the trunk or limbs, and Lhermitte sign (an electric shock–like sensation along the spine precipitated by neck flexion) may be present. Physical examination may show low-grade fever, glossitis, lemon-yellow discoloration of the skin, and cutaneous hyperpigmentation. Cerebral involvement produces confusion, depression, agitation, or psychosis with hallucinations. Spinal cord involvement is manifested by impaired vibratory and joint position sense, sensory gait ataxia, and spastic paraparesis with extensor plantar responses. Associated peripheral nerve involvement may cause loss of tendon reflexes in the legs and urinary retention.
Laboratory Findings
Hematologic abnormalities (Figure 4-9) include macro-cytic anemia, leukopenia with hypersegmented neutrophils, and thrombocytopenia with giant platelets. Because folate deficiency can produce identical changes, the diagnosis must be confirmed by the serum vitamin B12 level. When this is low (<100 pg/mL), a Schilling test determines whether defective intestinal absorption of vitamin B12 (as in pernicious anemia) is the cause. In pernicious anemia, the urinary excretion of orally administered vitamin B12 is abnormally low, and this abnormality can be corrected by coadministration of intrinsic factor. Diagnosis may be difficult when cerebral symptoms occur without anemia or spinal cord disease, requiring that the serum vitamin B12 level be determined routinely in patients with cognitive disorders, myelopathy, or peripheral neuropathy, regardless of whether anemia is present. T1-weighted MRI may show gadolinium enhancement of the posterior cord in B12 myelopathy (Figure 10–9) and deep T2-signal abnormalities in B12 encephalopathy, which resolve with treatment.
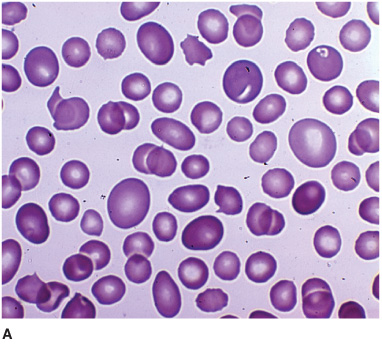
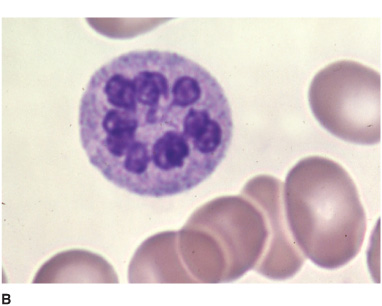
Figure 4-9. Peripheral blood smear from a patient with vitamin B12 deficiency showing oval macrocytes (A) and hypersegmented neutrophil (B). (From Kaushansky K, Lichtman M, Beutler E, Kipps T. Williams Hematology. 8th ed. New York: McGraw-Hill, 2010.)
Treatment
Treatment of neurologic manifestations is by prompt intramuscular administration of cyanocobalamin, as soon as blood is drawn to determine the serum vitamin B12 level. Daily injections are continued for 1 week, and a Schilling test is performed to determine the cause of deficiency. If, as in pernicious anemia, deficiency is not correctable by dietary supplementation or treatment of an underlying cause (eg, intestinal malabsorption), intramuscular vitamin B12 (typically 100 μg) is given at weekly intervals for several months and monthly thereafter. Alternatively, oral treatment with 100 to 250 μg/d may be possible, because even with impaired absorption, this can provide the required 5 μg/d. The reversibility of neurologic complications depends on their duration, and abnormalities present for more than 1 year are less likely to resolve with treatment. Encephalopathy may begin to clear within 24 hours after the first vitamin B12 dose, but full neurologic recovery, when it occurs, may take several months.
ORGAN SYSTEM FAILURE
HEPATIC ENCEPHALOPATHY
Hepatic encephalopathy occurs as a complication of cirrhosis, portosystemic shunting, chronic active hepatitis, or fulminant hepatic necrosis after viral hepatitis. Alcoholism is the most common underlying disorder. The syndrome may be chronic and progressive or acute in onset; in the latter case, gastrointestinal hemorrhage, systemic infection, dehydration, and sedative drugs are frequent precipitating factors. Liver disease produces cerebral symptoms by impairing hepatocellular detoxifying mechanisms or by the portosystemic shunting of venous blood. As a result, ammonia and perhaps other nitrogenous toxins accumulate in the blood and diffuse into the brain, where they appear to cause astrocyte swelling and brain edema.
Clinical Findings
Symptoms of encephalopathy may precede systemic symptoms such as nausea, anorexia, and weight loss. Recent gastrointestinal bleeding, consumption of high-protein foods, use of sedatives or diuretics, or systemic infection may cause clinical decompensation.
Physical examination may reveal systemic signs of liver disease. Cognitive disturbances include somnolence, agitation, and coma. Ocular reflexes are usually brisk. Nystagmus, tonic downward ocular deviation, and disconjugate eye movements may be seen. The most helpful neurologic sign of a metabolic disturbance, although not restricted to liver disease, is asterixis (Figure 4-10)—a flapping tremor of the outstretched, dorsiflexed hands or feet that results from impaired postural control. Other motor abnormalities include tremor, myoclonus, paratonic rigidity, spasticity, decorticate or decerebrate posturing, and extensor plantar responses. Focal neurologic signs and focal or generalized seizures may occur.
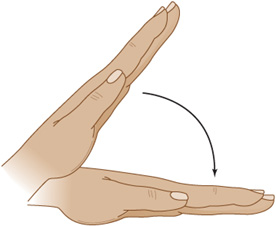
Figure 4-10. Asterixis, a flapping tremor of the outstretched hands or feet, is often associated with hepatic encephalopathy, but can be seen in a variety of metabolic disorders.
Laboratory Findings
Laboratory studies may show elevated serum bilirubin, transaminases, ammonia, prothrombin time (PT) and partial thromboplastin time (PTT), and respiratory alkalosis. The most specific CSF abnormality is elevated glutamine (Figure 4-11). The electroencephalogram (EEG) may be diffusely slow with triphasic waves.
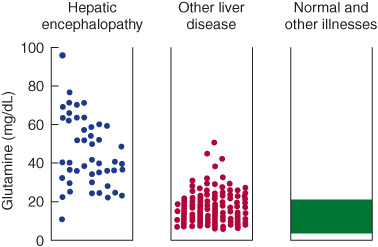
Figure 4-11.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


