weakness in previously involved or seemingly uninvolved muscles. Muscle pain and ease of fatigue are common. Slow progression occurs and may lead to increasing restriction of daily activities. The postpolio syndrome probably relates to loss of anterior horn cells with aging from a pool that was depleted by the original infection. There is no specific treatment.
WEST NILE VIRUS INFECTION
West Nile virus infection is acquired from infected mosquitos. Its most common manifestation is meningoencephalitis. Acute paralytic poliomyelitis is another manifestation and is characterized by acute, focal or generalized, asymmetric weakness or by a rapidly ascending quadriplegia that may be mistaken for the Guillain-Barré syndrome. Electrodiagnostic studies may be helpful in showing the nature and extent of involvement, distinguishing the disorder from a neuropathy, and guiding prognosis. Examination of the CSF is also helpful; there is a pleocytosis, often with a predominance of neutrophils, and viral-specific IgM antibodies are also found. Treatment is supportive, as in paralytic polio virus infection.
NERVE ROOT AND PLEXUS LESIONS
ACUTE INTERVERTEBRAL DISK PROLAPSE
LUMBAR DISK PROLAPSE
Acute prolapse of a lumbar disk (Figure 9-9) generally leads to pain in the back and in a radicular distribution (L5 or S1) in the leg, where it is often accompanied by numbness and paresthesias (Table 9-12). A motor deficit also may be found, depending on the root affected. An L5 radiculopathy causes weakness of dorsiflexion of the foot and toes, whereas S1 root involvement produces weakness of plantar flexion of the foot and a depressed ankle jerk. Movement of the spine is restricted, and there is local back tenderness and palpable spasm of the paraspinous muscles. Straight-leg raising in the supine position is restricted, often to approximately 20 or 30 degrees of hip flexion, from a normal value of approximately 80 or 90 degrees, because of reflex spasm of the hamstring muscles (Lasègue sign). A centrally prolapsed disk can lead to bilateral symptoms and signs and to sphincter involvement. The symptoms and signs of a prolapsed lumbar intervertebral disk can be either sudden or insidious in onset and may follow trauma. Pelvic and rectal examination and plain x-rays of the spine help to exclude lesions such as tumors.
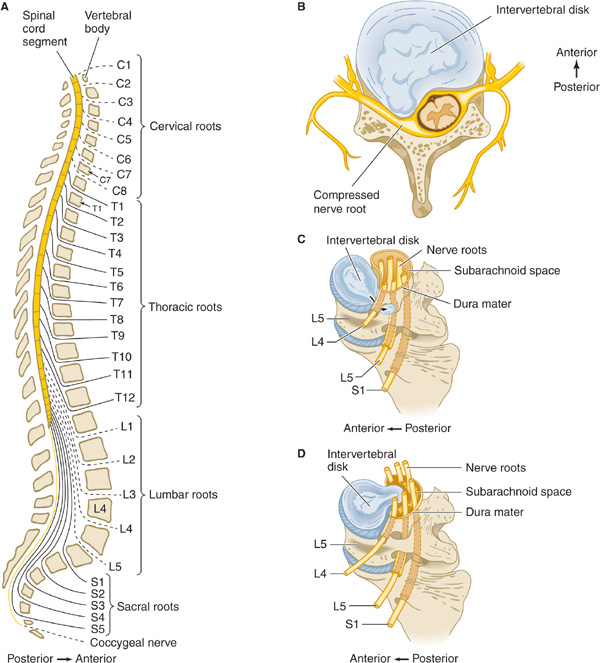
Figure 9-9. A. Lateral view of the vertebral column, showing the levels at which the various nerve roots exit; nerves exit above their numbered vertebral body in the cervical spine but below in the lumbar spine. B. Lateral disk prolapse in cervical spine, causing compression of exiting nerve root and compressing cervical cord. C. Lateral disk prolapse in lumbar spine, causing compression of the root exiting at the next lower vertebral level (eg, L4 disk compresses the L5 nerve root). D. Central disk prolapse in lumbar spine, causing bilateral root compression.
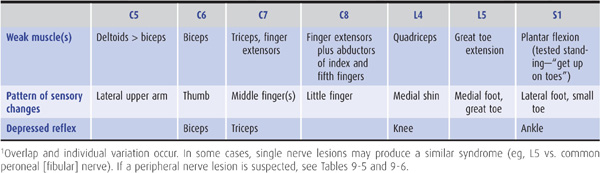
Table 9-12. The most common patterns of weakness, sensory symptoms, and reflex changes in nerve root lesions.1
Bed rest on a firm mattress for 2 or 3 days followed by gradual mobilization often permits symptoms to settle, but persisting pain, an increasing neurologic deficit, or any evidence of sphincter dysfunction should lead to CT, MRI, or CT myelography, followed by surgical treatment. Drug treatment for pain includes aspirin or acetaminophen with 30 mg of codeine, two doses three or four times daily, or other nonsteroidal analgesics such as ibuprofen or naproxen. Muscle spasm may respond to cyclobenzaprine 10 mg orally three times daily or as needed and tolerated, or diazepam 5 to 10 mg orally three times daily or as tolerated.
CERVICAL DISK PROLAPSE
Acute protrusion of a cervical disk can occur at any age, often with no preceding trauma, and leads to pain in the neck and radicular pain in the arm. The pain is exacerbated by head movement. With lateral herniation of the disk, a motor, sensory, or reflex deficit may be found in a radicular (usually C6 or C7) distribution on the affected side (Table 9-12); with more centrally directed herniations, the spinal cord may also be involved (Figure 9-9), leading to a spastic paraparesis and sensory disturbance in the legs, sometimes accompanied by impaired sphincter function. The diagnosis is confirmed by CT scanning, MRI, or CT myelography. Surgical treatment may be needed.
CERVICAL SPONDYLOSIS
This disorder has been described earlier as a cause of myelopathy.
TRAUMATIC AVULSION OF NERVE ROOTS
ERB-DUCHENNE PARALYSIS
Traumatic avulsion of the C5 and C6 roots can occur at birth as a result of traction on the head during delivery of the shoulder. It can also be the result of injuries causing excessive separation of the head and shoulder. It leads to loss of shoulder abduction and elbow flexion. In consequence, the affected arm is held internally rotated at the shoulder, with a pronated forearm and extended elbow. The biceps and brachioradialis jerks are lost, but sensory impairment is usually inconspicuous, because it is confined to a small area overlying the deltoid muscle.
KLUMPKE PARALYSIS
Involvement of the C8 and T1 roots causes paralysis and wasting of the small muscles of the hand and of the long finger flexors and extensors. Horner syndrome is sometimes an associated finding. This kind of lower plexus paralysis often follows a fall that has been arrested by grasping a fixed object with one hand or may result from traction on the abducted arm.
BRACHIAL PLEXOPATHY
NEURALGIC AMYOTROPHY (IDIOPATHIC BRACHIAL PLEXOPATHY)
This disorder, also referred to as Parsonage-Turner syndrome, typically begins with severe pain about the shoulder followed within a few days by weakness, reflex changes, and sensory disturbances in the arm, often involving the C5 and C6 segments especially. Symptoms and signs are unilateral in approximately 70% of patients. The motor deficit sometimes corresponds to the territory of an individual nerve, especially the axillary, suprascapular, or radial nerve, but in other instances appears to arise in the brachial plexus (Figure 9-10). Wasting of the affected muscles is often profound. Recurrences occur in approximately 25% of patients.
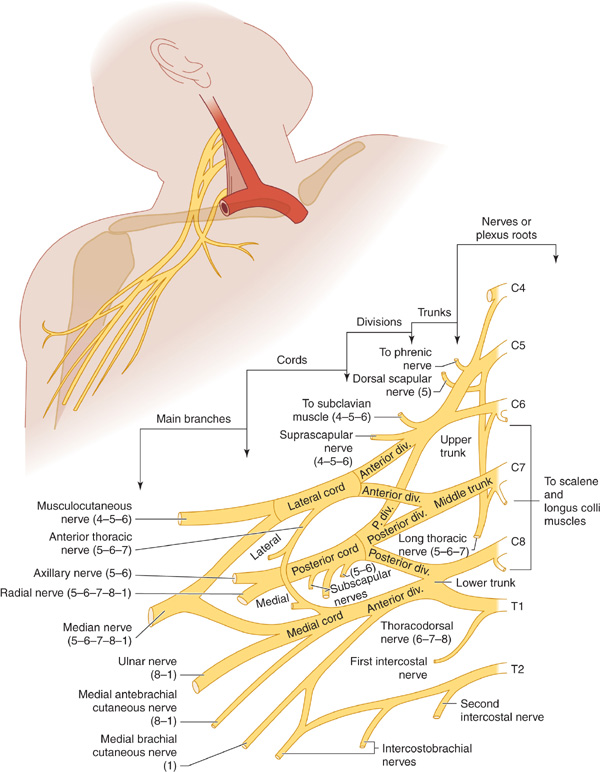
Figure 9-10. Brachial plexus. The numbers in parentheses refer to the segmental origin of the nerves depicted. (Reproduced from Waxman SG. Clinical Neuroanatomy. 26th ed. New York, NY:McGraw-Hill; 2010.)
Treatment is symptomatic. The combination of a non-steroidal anti-inflammatory drug and an opiate is often effective treatment for pain in the acute phase, whereas persisting pain may respond to an anticonvulsant or tricyclic antidepressant. Recovery over the ensuing weeks and months is the rule—but it is sometimes incomplete.
CERVICAL RIB SYNDROME
The C8 and T1 roots or the lower trunk of the brachial plexus may be compressed by a cervical rib or band arising from the seventh cervical vertebra. This leads to weakness and wasting of intrinsic hand muscles, especially those in the thenar eminence, accompanied by pain and numbness in the appropriate dermatomal distribution (often like that of an ulnar nerve lesion but extending up the medial border of the forearm). The subclavian artery may also be compressed; this forms the basis of Adson test for diagnosing the disorder. The radial pulse decreases in amplitude when the seated patient turns the head to the affected side and inhales deeply. A positive Adson test, however, also can be seen in normal subjects; a supraclavicular bruit during the maneuver supports the diagnosis of subclavian artery compromise.
X-rays may show the cervical rib or a long transverse process of the seventh cervical vertebra, but normal findings do not exclude the possibility of a cervical band. Electromyography shows evidence of chronic partial denervation in the hand—in a territory beyond that of any individual peripheral nerve. Nerve conduction studies show no evidence of peripheral nerve disease, but there is a small or absent ulnar sensory nerve action potential on stimulation of the little finger. Treatment is by surgical excision of the rib or band.
OTHER CAUSES OF BRACHIAL PLEXOPATHY
Brachial plexopathy may occur in patients with neoplastic infiltration (especially by breast or lung cancer) or as a consequence of radiation therapy, following median sternotomy, and after trauma. Electrophysiologic studies are important in defining the extent and severity of involvement and localizing the lesion. The absence of electrodiagnostic abnormalities despite an adequate examination suggests an incorrect diagnosis and raises the possibility of conversion reactions, malingering, or so-called nonneurogenic thoracic outlet syndrome (a controversial and disputed entity).
LUMBOSACRAL PLEXOPATHY
A disorder similar to idiopathic brachial plexopathy occasionally affects the lumbosacral plexus. Treatment is symptomatic. As noted previously for brachial plexopathy, lumbosacral plexopathy can also be caused by neoplasms (colorectal or gynecologic cancers, sarcomas, or lymphomas) or radiation. Intrapartum maternal lumbosacral plexopathy is an uncommon but important cause of acute foot drop developing during labor. It occurs mostly in short women and relates to compression of the lumbosacral trunk by the fetal head at the pelvic brim. Most patients recover completely within 6 months.
DISORDERS OF PERIPHERAL NERVE
The term peripheral neuropathy designates a disturbance in function of one or more peripheral nerves. Several types of peripheral neuropathy are distinguishable by the extent of involvement.
Depending on the underlying cause, there may be selective involvement of motor, sensory, or autonomic fibers or more diffuse involvement of all fibers in the peripheral nerve.
The clinical deficit is usually a mixed one, and sensory symptoms and signs are often the initial—and most conspicuous—feature of peripheral nerve involvement. Further discussion of these disorders and their treatment is therefore deferred to Chapter 10, except in those instances in which presentation is typically with acute motor deficits. For convenience, however, the root and peripheral nerve supply of the major limb muscles is set forth in Tables 9-5 and 9-6. Reference to the tables should facilitate evaluation of patients presenting with focal weakness of lower motor neuron type.
POLYNEUROPATHY
In polyneuropathy, because there is symmetric and simultaneous involvement of various nerves, the deficits resulting from individual nerves cannot be recognized clinically. Polyneuropathies are discussed in Chapter 10, but brief mention is made here of those neuropathies in which patients present with acute weakness.
ACUTE INFLAMMATORY POLYRADICULONEUROPATHY (GUILLAIN-BARRÉ SYNDROME)
This disorder commonly presents with weakness that is often symmetric and most often begins in the legs. The speed and extent of progression vary, but in severe cases there is marked weakness of all limbs in addition to bilateral facial weakness. There may also be subjective sensory complaints, although objective sensory disturbances are usually far less conspicuous than motor deficits. Autonomic involvement is common and may lead to a fatal outcome, as may aspiration pneumonia or impaired respiration from weakness. Further details about this disorder are given in Chapter 10.
CRITICAL ILLNESS POLYNEUROPATHY
Patients with sepsis and multiorgan failure may develop a polyneuropathy that often first comes to attention when unexpected difficulty is encountered in weaning the patients from a mechanical ventilator. In more advanced cases, wasting and weakness of the extremities are present and the tendon reflexes are lost. Sensory abnormalities are overshadowed by the motor deficit.
Electrophysiologic studies are helpful in distinguishing the disorder from Guillain-Barré syndrome by revealing an axonal neuropathy. The underlying pathogenesis is obscure, but the polyneuropathy may relate in part to the use of neuromuscular blocking agents or corticosteroids. Treatment is supportive, with the long-term outlook being good in patients who recover from the underlying critical illness.
DIPHTHERITIC POLYNEURITIS
Infection with Corynebacterium diphtheriae can occur either in the upper respiratory tract or by infection of a skin wound, and neuropathy results from a neurotoxin that is released by the organism. Diphtheria toxin kills cells by inactivating eukaryotic elongation factor-2 and thereby blocking protein synthesis.
Palatal weakness may develop 2 to 3 weeks after infection of the throat, and cutaneous diphtheria may be followed by focal weakness of neighboring muscles after a similar interval. Impaired pupillary responses to accommodation may occur approximately 4 to 5 weeks after infection and a generalized sensorimotor polyneuropathy after 1 to 3 months. The polyneuropathy may follow a biphasic course, with further deterioration occurring 5 to 6 weeks after onset. The weakness may be asymmetric and is often more marked proximally than distally. The tendon reflexes may be depressed or absent. Respiratory paralysis occurs in severe cases. Recovery usually occurs over the following 2 to 3 months but may take longer in severe cases.
In patients with diphtheritic polyneuritis, CSF protein content is usually increased, and there may be a mild pleocytosis. Electrophysiologic studies show a slowing of nerve conduction velocity, but this is often not manifest until the patient has begun to improve clinically. Treatment consists of early administration of equine diphtheria antitoxin without awaiting the results of bacterial culture, provided the patient is not hypersensitive to horse serum. A 2-week course of penicillin or erythromycin will usually eradicate the infection but does not alter the incidence of serious complications. In patients with marked weakness, supportive measures, including ventilatory assistance, are necessary.
PARALYTIC SHELLFISH POISONING
Mussels and clams found on the East and West Coasts of the United States may be dangerous to eat, especially in the summer months. They feed on poisonous varieties of plankton and come to contain saxitoxin, which blocks sodium channels—and therefore action potentials—in motor and sensory nerves and in muscle. A rapidly progressive acute peripheral neuropathy, with sensory symptoms and a rapidly ascending paralysis, begins within 30 minutes after eating affected shellfish and may lead to respiratory paralysis and death. There is no available antitoxin, but with proper supportive care (including mechanical ventilation if necessary) the patient recovers completely. A cathartic or enema may help remove unabsorbed toxin.
PORPHYRIA
Acute polyneuropathy may occur with the hereditary hepatic porphyrias. Attacks can be precipitated by drugs (eg, barbiturates, estrogens, sulfonamides, griseofulvin, phenytoin, and succinimides) that can induce the enzyme δ-aminolevulinic acid synthetase, or by infection, a period of fasting, or, occasionally, menses or pregnancy. They usually last for 1 to 2 weeks but can be life-threatening.
Colicky abdominal pain—sometimes also felt in the back or thighs—frequently precedes neurologic involvement, and there may also be anxiety, agitation, acute confusion or delirium, and convulsions. Weakness is the major neurologic manifestation and is due to a predominantly motor polyneuropathy that causes a symmetric disturbance that is sometimes more marked proximally than distally. It may begin in the upper limbs and progress to involve the lower limbs or trunk. Progression occurs at a variable rate and can lead to complete flaccid quadriparesis with respiratory paralysis over a few days. Sensory loss occurs also but is less conspicuous and extensive; muscle pain is sometimes prominent. The tendon reflexes may be depressed or absent. Acute attacks may be accompanied by fever, excessive sweating, persistent tachycardia, hypertension, hyponatremia (attributed to inappropriate secretion of antidiuretic hormone), and peripheral leukocytosis, and patients may become dehydrated.
The CSF may show a slight increase in protein concentration and a slight pleocytosis. The diagnosis is confirmed by demonstrating increased levels of porphobilinogen and δ-aminolevulinic acid in the urine or deficiency of porphobilinogen deaminase in red blood cells (acute intermittent porphyria) or of coproporphyrinogen oxidase in lymphocytes (hereditary coproporphyria).
Treatment is with intravenous dextrose to suppress the heme biosynthetic pathway and propranolol to control tachycardia and hypertension. Hematin (4 mg/kg by intravenous infusion over 15 minutes once daily) is also effective in improving the clinical state. The best index of progress is the heart rate. The abdominal and mental symptoms (but not the neuropathy) may be helped by chlorpromazine or another phenothiazine. Pain relief may require opiates. Patients with impaired respiratory function, depressed level of consciousness, or convulsions should be followed in an intensive care unit. Respiratory failure may necessitate tracheostomy and mechanical ventilation. Recovery from paralysis is gradual and may not be complete.
Any precipitant should be removed: Precipitating medications should be discontinued, infections should be treated, and inadequate diets corrected. Preventing future acute attacks by avoiding known precipitants is important. Identification of the responsible genetic mutation in an affected patient allows for genetic screening of other family members to prevent acute attacks in those with occult disease. Different genes have been implicated in different porphyrias: In acute intermittent porphyria, the responsible mutation is in the porphobilinogen deaminase (PBGD) gene.
ACUTE ARSENIC OR THALLIUM POISONING
Acute arsenic or thallium poisoning can produce a rapidly evolving sensorimotor polyneuropathy, often with an accompanying or preceding gastrointestinal disturbance and crampy abdominal pain. Arsenic may also cause a skin rash, with increased skin pigmentation and marked exfoliation, together with the presence of Mees lines (transverse white lines) on the nails in long-standing cases. Thallium can produce a scaly rash and hair loss. Sensory symptoms, often painful, are usually the earliest manifestation of polyneuropathy; this is followed by symmetric motor impairment, which is usually more marked distally than proximally and occurs in the legs rather than the arms.
The CSF protein may be increased, with little or no change in cell content, and the electrophysiologic findings sometimes resemble those of Guillain-Barré syndrome, especially in the acute phase of the disorder. The diagnosis of arsenic toxicity is best established by measuring the arsenic content of hair protected from external contamination (eg, hair from the pubic region). Urine also contains arsenic in the acute phase. The diagnosis of thallium poisoning is made by finding thallium in body tissues or fluids, especially in urine. The degree of neurologic recovery depends on the severity of the intoxication. Chelating agents are of uncertain value.
ORGANOPHOSPHATE POLYNEUROPATHY
Organophosphate compounds are widely used as insecticides and are also the active principles in the nerve gas of chemical warfare. They have a variety of acute toxic effects, particularly manifestations of cholinergic crisis caused by inhibition of acetylcholinesterase. Some organophosphates, however, also induce a delayed polyneuropathy that generally begins approximately 1 to 3 weeks after acute exposure.
Cramping muscle pain in the legs is usually the initial symptom of neuropathy, sometimes followed by distal numbness and paresthesias. Progressive leg weakness then occurs, along with depression of the tendon reflexes. Similar deficits may develop in the upper limbs after several days. Sensory disturbances also develop in some instances, initially in the legs and then in the arms, but these disturbances are often mild or inconspicuous.
Examination shows a distal, symmetric, predominantly motor polyneuropathy, with wasting and flaccid weakness of distal leg muscles. In some patients, involvement may be severe enough to cause quadriplegia, whereas in others the weakness is much milder. Mild pyramidal signs also may be present. Objective evidence of sensory loss is usually slight.
The acute effects of organophosphate poisoning may be prevented by the use of protective masks and clothing. Treatment after exposure includes decontamination of the skin with bleach or soap and water and administration of atropine 2 to 6 mg every 5 minutes and pralidoxime 1 g every hour for up to 3 hours, both given intramuscularly or intravenously. Atropine blocks muscarinic cholinergic receptors, and pralidoxime binds to and reactivates acetylcholinesterase. There is no treatment for the neuropathy other than supportive care. Recovery of peripheral nerve function may occur with time, but central deficits are usually permanent and may govern the extent of ultimate functional recovery.
MONONEUROPATHY MULTIPLEX
This term signifies that there is involvement of various nerves but in an asymmetric manner and at different times, so that the individual nerves involved can usually be identified until the disorder reaches an advanced stage. Comment here will be restricted to two disorders characterized by motor involvement in the absence of sensory symptoms and signs.
LEAD TOXICITY
Lead toxicity is common among persons involved in the manufacture or repair of storage batteries or other lead-containing products, the smelting of lead or lead-containing ores, and the ship breaking industry. It may also occur in persons using lead-containing paints or those who ingest contaminated alcohol. Inorganic lead can produce dysfunction of both the central and peripheral nervous systems. In children, who can develop toxicity by ingesting lead-containing paints that flake off old buildings or furniture, acute encephalopathy is the major neurologic feature.
The peripheral neuropathy is predominantly motor, and in adults it is more severe in the arms than in the legs. It typically affects the radial nerves, although other nerves may also be affected, leading to an asymmetric progressive motor disturbance. Sensory loss is usually inconspicuous or absent. There may be loss or depression of tendon reflexes. Systemic manifestations of lead toxicity include anemia, constipation, colicky abdominal pain, gum discoloration, and nephropathy. The extent to which exposed workers develop minor degrees of peripheral nerve damage as a result of lead toxicity is not clear. Similarly, there is no agreement about the lowest concentration of blood lead that is associated with damage to the peripheral nerves.
The optimal approach to treatment is not known, but intravenous or intramuscular edetate calcium disodium (EDTA) and oral penicillamine have been used, as has dimercaprol (BAL).
MULTIFOCAL MOTOR NEUROPATHY
This disorder is characterized by progressive asymmetric wasting and weakness, electrophysiologic evidence of multifocal motor demyelination with partial motor conduction block but normal sensory responses, and the presence of antiglycolipid (usually anti-GM1 IgM) antibodies in the serum of many patients. Cramps and fasciculations sometimes occur and may lead to an erroneous diagnosis of motor neuron disease unless electrophysiologic studies are performed. There is no sensory loss or evidence of upper motor neuron involvement. The disorder typically has an insidious onset and chronic course, but variants with a more acute onset have been described. For a diagnosis to be made with confidence, the motor deficit should be in the distribution of two or more named nerves and related to conduction block outside of common entrapment sites. A variant with involvement of only a single nerve has been described (monofocal motor neuropathy). The conduction block is a major consequence of demyelination, but axonal excitability changes also contribute to conduction failure. Treatment with prednisone and plasmapheresis has been disappointing, but patients may improve after treatment with cyclophosphamide 1 g/m2 intravenously once per month for 6 months or in response to human immunoglobulin 2 g/kg intravenously given over 3 to 5 days. Improvement is sometimes associated with a decrease in anti-GM1 antibody levels.
MONONEUROPATHY SIMPLEX
In mononeuropathy simplex there is involvement of a single peripheral nerve. Most of the common mononeuropathies entail both motor and sensory involvement (as discussed in Chapter 10). Accordingly, only Bell palsy, which leads primarily to a motor deficit, is discussed here.
BELL PALSY
Facial weakness of the lower motor neuron type caused by idiopathic facial (VII) nerve involvement outside the central nervous system, without evidence of more widespread neurologic disease, has been designated Bell palsy. The cause is unclear, but the disorder occurs more commonly in pregnant women and diabetics. Increasing evidence suggests that reactivation of herpes simplex virus type 1 or varicella-zoster virus infection in the geniculate ganglion may injure the facial nerve and is responsible for Bell palsy in at least some patients.
Facial weakness is often preceded or accompanied by pain about the ear. Weakness generally comes on abruptly but may progress over several hours or even a day or so. Depending on the site of the lesion, there may be associated impairment of taste, lacrimation, or hyperacusis. There may be paralysis of all muscles supplied by the affected nerve (complete palsy) or variable weakness in different muscles (incomplete palsy). Clinical examination reveals no abnormalities beyond the territory of the facial nerve.
Most patients recover completely without treatment, but this may take several days in some instances and several months in others. A poor prognosis for complete recovery is suggested by severe pain at onset and complete palsy when the patient is first seen. Even if recovery is incomplete, permanent disfigurement or some other complication affects only about 10% of patients.
Other conditions that can produce facial palsy include tumors, herpes zoster infection of the geniculate ganglion (Ramsay Hunt syndrome), Lyme disease, AIDS, sarcoidosis, or any inflammatory process involving the subarachnoid space, such as infective or neoplastic meningitis. Facial palsies that are bilateral or are associated with another cranial neuropathy merit lumbar puncture and brain MRI to search for an underlying cause.
DISORDERS OF NEUROMUSCULAR TRANSMISSION
MYASTHENIA GRAVIS
PATHOGENESIS
Myasthenia gravis is caused by variable block of neuromuscular transmission related to an immune-mediated decrease in the number of functioning nicotinic acetylcholine receptors (Figure 9-11). In approximately 80% of cases, antibodies to the skeletal muscle nicotinic acetylcholine receptor are present and lead to loss of receptor function. In patients seronegative for these antibodies, the disease is probably also immune mediated; many of these patients have antibodies against the muscle-specific receptor tyrosine kinase (MuSK) that is involved in the clustering of acetylcholine receptors during development and is also expressed in mature neuromuscular junctions. A similar disorder can occur in patients receiving penicillamine for rheumatoid arthritis; it frequently remits when the drug is discontinued.
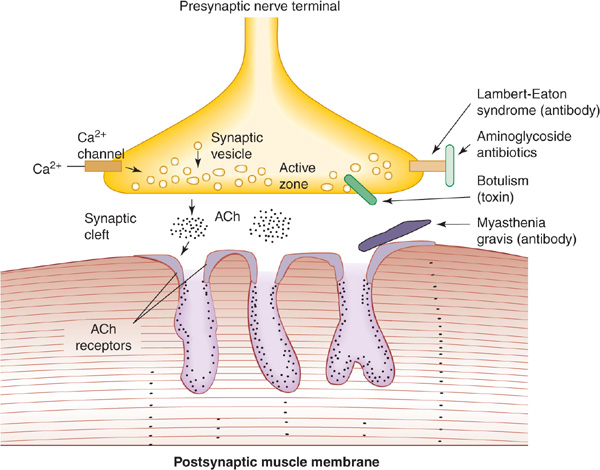
Figure 9-11. Sites of involvement in disorders of neuromuscular transmission. At left, normal transmission involves depolarization-induced influx of calcium (Ca) through voltage-gated channels. This stimulates release of acetylcholine (ACh) from synaptic vesicles at the active zone and into the synaptic cleft. ACh binds to ACh receptors and depolarizes the postsynaptic muscle membrane. At right, disorders of neuromuscular transmission result from blockage of Ca channels (Lambert-Eaton syndrome or aminoglycoside antibiotics), impairment of Ca-mediated ACh release (botulinum toxin), or antibody-induced internalization and degradation of ACh receptors (myasthenia gravis).
CLINICAL FINDINGS
Myasthenia gravis can occur at any age and is sometimes associated with thymic tumor, thyrotoxicosis, rheumatoid arthritis, or disseminated lupus erythematosus. More common in females than males, it is characterized by fluctuating weakness and easy fatigability of voluntary muscles; muscle activity cannot be maintained, and initially powerful movements weaken readily. There is a predilection for the external ocular muscles and certain other cranial muscles, including the masticatory, facial, pharyngeal, and laryngeal muscles. Respiratory and limb muscles may also be affected.
History
Although the onset of the disease is usually insidious, the disorder is sometimes unmasked by a concurrent infection, which leads to an exacerbation of symptoms. Exacerbations may also occur in pregnancy or before the menstrual periods. Symptoms may be worsened by quinine, quinidine, procainamide, propranolol, phenytoin, lithium, tetracycline, and aminoglycoside antibiotics, which therefore should be avoided in such patients.
Myasthenia follows a slowly progressive course. Patients present with ptosis, diplopia, difficulty in chewing or swallowing, nasal speech, respiratory difficulties, or weakness of the limbs (Table 9-13). These symptoms often fluctuate in intensity during the day, and this diurnal variation is superimposed on longer-term spontaneous relapses and remissions that may last for weeks.
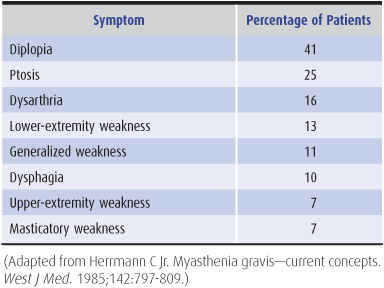
Table 9-13. Presenting symptoms in myasthenia gravis.
Examination
Clinical examination confirms the weakness and fatigability of affected muscles. The weakness does not conform to the distribution of any single nerve, root, or level of the central nervous system. In more than 90% of cases the extraocular muscles are involved, leading to often asymmetric ocular palsies and ptosis. Pupillary responses are not affected. The characteristic feature of the disorder is that sustained activity of affected muscles leads to temporarily increased weakness. Thus sustained upgaze for 2 minutes can lead to increased ptosis, with power in the affected muscles improving after a brief rest. In advanced cases, there may be some mild atrophy of affected muscles. Sensation is normal, and there are usually no reflex changes.
DIAGNOSIS
The diagnosis of myasthenia gravis can generally be confirmed by the benefit that follows administration of anti-cholinesterase drugs; the power of affected muscles is influenced at a dose that has no effect on normal muscles and slight, if any, effect on muscles weakened by other causes.
The most commonly used pharmacologic test is the edrophonium (Tensilon) test. Edrophonium is given intravenously in a dose of 10 mg (1 mL), of which 2 mg is given initially as a test dose and the remaining 8 mg approximately 30 seconds later if the test dose is well tolerated. In myasthenic patients, there is an obvious improvement in the strength of weak muscles that lasts for approximately 5 minutes. Alternatively, 1.5 mg of neostig-mine can be given intramuscularly, with a response that lasts for about 2 hours. Atropine sulfate (0.6 mg intravenously) should be available to counteract the muscarinic cholinergic side effects of increased salivation, diarrhea, and nausea. Atropine does not affect nicotinic cholinergic function at the neuromuscular junction. The longer-acting neostigmine reduces the incidence of false-negative evaluations.
INVESTIGATIVE STUDIES
X-rays and CT scans of the chest, with and without contrast, may reveal a coexisting thymoma. Normal studies do not exclude this possibility. Impaired neuromuscular transmission can be detected electrophysiologically by a decremental response of muscle to repetitive supramaximal stimulation (at 2 or 3 Hz) of its motor nerve, but normal findings do not exclude the diagnosis. Single-fiber electro-myography shows increased variability in the interval between two muscle fiber action potentials from the same motor unit in clinically weak muscles. Measuring serum acetylcholine receptor antibody levels is often helpful, because increased values are found in 80% to 90% of patients with generalized myasthenia gravis.
TREATMENT
Medications (referred to earlier) that impair neuromuscular transmission should be avoided. The following approaches to treatment are recommended.
Anticholinesterase Drugs
Treatment with these drugs provides symptomatic benefit without influencing the course of the underlying disease. The mainstay of treatment is pyridostigmine, at doses individually determined but usually between 30 and 180 mg (average, 60 mg) four times daily. The older drug neostigmine may still be used, in rare instances, by parenteral administration. Small doses of atropine may attenuate side effects such as bowel hypermotility or hypersalivation. Overmedication can lead to increased weakness, which, unlike myasthenic weakness, is unaffected or enhanced by intravenous edrophonium. Such a cholinergic crisis may be accompanied by pallor, sweating, nausea, vomiting, salivation, colicky abdominal pain, and miosis.
Thymectomy
Thymectomy should be performed in patients younger than 60 years of age, and considered in those older, with weakness that is not restricted to the extraocular muscles. Although thymectomy usually leads to symptomatic benefit or remission, the mechanism by which it confers benefit is unclear, and its beneficial effect may not be evident immediately.
Corticosteroids
Corticosteroids are indicated for patients who have responded poorly to anticholinesterase drugs and have already undergone thymectomy. Treatment is initiated with the patient in the hospital, as weakness may initially be exacerbated. An initial high dose of prednisone (60-100 mg/d orally) can be tapered gradually to a relatively low maintenance level (5-15 mg/d) as improvement occurs. Alternate-day treatment is helpful in reducing the incidence of side effects, which are described (as clinical findings) in the section on hyperadrenalism (Cushing syndrome) in Chapter 4.
Azathioprine
This drug can be used in patients with severe or progressive disease despite thymectomy and treatment with anticholinesterases and corticosteroids. It can also be given in place of high doses of corticosteroids to patients who show no sustained benefit with low doses. The usual dose is 2 to 3 mg/kg/d, increased from a lower initial dose.
Plasmapheresis
Plasmapheresis may be used to achieve temporary improvement in patients deteriorating rapidly or in myasthenic crisis and in certain special circumstances, such as before surgery that is likely to produce postoperative respiratory compromise.
Intravenous Immunoglobulins
Intravenous immunoglobulins also have been used to provide temporary benefit in circumstances similar to those in which plasmapheresis is used.
Mycophenolate Mofetil
This agent selectively inhibits proliferation of T and B lymphocytes and has been used as an immunosuppressant with only modest side effects, including diarrhea, nausea, abdominal pain, fever, leukopenia, and edema. Several studies indicate that many patients with myasthenia gravis improve or are able to lower their steroid intake in response to this medication (unlabeled use; 1 g twice daily by mouth), but usually after a delay of several months.
PROGNOSIS
Most patients can be managed successfully with drug treatment. The disease may have a fatal outcome because of respiratory complications such as aspiration pneumonia.
MYASTHENIC SYNDROME (LAMBERT-EATON SYNDROME)
PATHOGENESIS
This disorder has a well-recognized association with an underlying neoplasm and occasionally may be associated with such autoimmune diseases as pernicious anemia; occasionally no cause is found. In the paraneoplastic disorder, antibodies directed against tumor antigens cross-react with voltage-gated calcium channels involved in acetylcho-line release, leading to a presynaptic disturbance of neuro-muscular transmission (Figure 9-11).
CLINICAL FINDINGS
Clinically there is weakness, especially of the proximal muscles of the limbs. Unlike myasthenia gravis, however, the extraocular muscles are characteristically spared, and power steadily increases if a contraction is maintained. Autonomic disturbances, such as dry mouth, constipation, and impotence, may also occur.
DIAGNOSIS
The diagnosis is confirmed electrophysiologically by the incremental response to repetitive nerve stimulation. There is a remarkable increase in the size of the muscle response to stimulation of its motor nerve at high rates—even in muscles that are not clinically weak. The presence of autoantibodies to the P/Q subtype of voltage-gated calcium channels, found on the presynaptic membrane of the neuromuscular junction, is highly sensitive and specific to the Lambert-Eaton syndrome of any etiology.
TREATMENT
Immunosuppressive drug therapy (corticosteroids and azathioprine as described earlier for myasthenia gravis) and plasmapheresis or intravenous immunoglobulin therapy may lead to improvement. Guanidine hydrochloride (25-50 mg/kg/d in three or four divided doses) is sometimes helpful in seriously disabled patients, but adverse effects of the drug include bone marrow suppression and renal failure. The response to treatment with anticholinest-erase drugs such as pyridostigmine or neostigmine, alone or in combination with guanidine, is variable but usually disappointing. 3,4-Diaminopyridine (amifampridine), a potassium channel antagonist that enhances the release of acetylcholine at the neuromuscular junction, used in doses up to 25 mg orally four times daily, may improve weakness and autonomic dysfunction; paresthesia is a common side effect, and seizures may occur. The disease improves with treatment of the underlying condition, often a small-cell lung cancer.
BOTULISM
PATHOGENESIS
The toxin of Clostridium botulinum can cause neuromuscular paralysis. It acts by preventing the release of acetylcho-line at neuromuscular junctions and autonomic synapses (Figure 9-11). Botulism occurs most commonly after ingestion of home-canned food that is contaminated with the toxin; it occurs rarely from infected wounds. The shorter the latent period between ingestion of the toxin and the onset of symptoms, the greater the dose of toxin and the risk for further involvement of the nervous system.
CLINICAL FINDINGS
Fulminating weakness begins 12 to 72 hours after ingestion of the toxin and characteristically is manifested by diplopia, ptosis, facial weakness, dysphagia, nasal speech, and then difficulty with respiration; weakness usually appears last in the limbs. In addition to the motor deficit, blurring of vision is characteristic, the pupils being dilated and unreactive, and there may be dryness of the mouth, paralytic ileus, and postural hypotension. There is no sensory deficit, and the tendon reflexes are usually unchanged unless the involved muscles are quite weak. Symptoms can progress for several days after their onset.
In infants, enteric infection with local production of the toxin leads to a different clinical picture with hypotonia, constipation, progressive weakness, and a poor suck. This is now the most common form of botulism in the United States.
INVESTIGATIVE STUDIES
Once the diagnosis is suspected, the local health authority should be notified and samples of the patient’s serum and the contaminated food (if available) sent to be assayed for toxin. The most common types of toxin encountered clinically are A, B, and E. Electrophysiologic studies may help confirm the diagnosis, as the evoked muscle response tends to increase in size progressively with repetitive stimulation of motor nerves at fast rates.
TREATMENT
Patients should be hospitalized, because respiratory insufficiency can develop rapidly and necessitates ventilatory assistance. Treatment with trivalent antitoxin (ABE) is commenced once it is established that the patient is not allergic to horse serum, but the effect on the course of the disease is unclear.
In wound botulism, antibiotic therapy is often prescribed but is of unclear benefit; either penicillin G (3 million units IV every 4 hours in adults) or metronidazole (500 mg IV every 8 hours in patients allergic to penicillin) is typically administered, but the regimen may need to be altered depending on the results of wound culture.
Guanidine hydrochloride (25-50 mg/kg/d in divided doses), a drug that facilitates release of acetylcholine from nerve endings, is sometimes helpful in improving muscle strength; anticholinesterase drugs are generally of no value.
In infants, human-derived botulinum immune globulin should be given intravenously as soon as possible in the course of the illness. Nursing and supportive care are important.
AMINOGLYCOSIDE ANTIBIOTICS
Large doses of antibiotics such as kanamycin and gentamicin can produce a clinical syndrome rather like botulism, because the release of acetylcholine from nerve endings is prevented. This effect may be related to calcium channel blockade (Figure 9-11). Symptoms resolve rapidly as the responsible drug is eliminated from the body. Note that these antibiotics are particularly dangerous in patients with preexisting disturbances of neuromuscular transmission and are therefore best avoided in patients with myasthenia gravis.
MYOPATHIC DISORDERS
MUSCULAR DYSTROPHIES
The muscular dystrophies are a group of inherited myopathic disorders characterized by progressive muscle weakness and wasting. They are subdivided by their mode of inheritance, age at onset, distribution of involved muscles, rate of progression, and long-term outlook (Table 9-14). A more satisfactory means of classification may be on genetic grounds. A number of genes have now been associated with the different muscular dystrophies (Table 9-15). These skeletal muscle genes encode sarcolemmal (eg, sarcoglycans), cytoskeletal (eg, dystrophin), cytosolic, extracellular matrix, and nuclear membrane proteins. Abnormalities of these proteins may lead to a greater susceptibility to necrosis of muscle fibers, but the molecular mechanisms involved are not yet clear. Genetic heterogeneity for the same phenotype has led to subdivision of the main clinical disorders, but the basis for the different clinical phenotypes is unknown.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


