Congenital and Childhood Central Nervous System Tumors
Congenital and Childhood Central Nervous System Tumors
James H. Garvin Jr
INTRODUCTION
Childhood brain tumors range from benign congenital lesions to aggressive, infiltrative cancers. Cure rates for malignant brain tumors are lower than those of most other common childhood cancers, and treatment sequelae may seriously compromise quality of survival. Nonetheless, research advances and refinement of treatment techniques continue to give a sense of cautious optimism. Current research focuses on validating experimental models and identifying molecular targets for novel therapies. An emerging concept is that malignant brain tumors are propagated from stem-like progenitor cells, which may be intrinsically resistant to standard radiation and chemotherapy but could be eliminated by targeted therapy with agents, which disrupt critical signaling pathways or induce differentiation.
EPIDEMIOLOGY
Malignant and benign central nervous system (CNS) tumors may arise at any time in infancy and childhood. Categorization as malignant or benign does not always reflect clinical behavior. Certain benign lesions such as craniopharyngioma can cause extensive damage, whereas low-grade astrocytomas, although classified as malignant, are rarely life-threatening. The peak age for malignant brain tumors is 3 to 4 years. Location is predominantly cerebral or ventricular in infants, cerebellar or brain stem in young children, and midbrain or cerebral in older children and adolescents. Astrocytomas are the largest group, mostly of low-grade histology; pilocytic astrocytomas (World Health Organization [WHO] grade I) account for 24% of all childhood brain tumors. Next in frequency are medulloblastomas, the most common high-grade malignant tumor (16%). After these are a diverse group of congenital and acquired lesions, including high-grade astrocytomas (14%), ependymomas (10%), craniopharyngiomas (5.6%), germ cell tumors (2.5%), and choroid plexus tumors (0.9%). Neuronal tumors (gangliocytoma) and mixed glioneuronal tumors (ganglioglioma, dysembryoplastic neuroepithelial tumor) are relatively uncommon. The principal primary brain tumors of adults (glioblastoma multiforme, meningioma, oligodendroglioma, pituitary adenoma) are rarely seen in children. Moreover, tumorigenesis of high-grade astrocytomas in children differs from that in adults, as deduced from comparative genomic hybridization studies. CNS metastases from solid tumors are also uncommon in children compared to adults. Cerebral metastases are occasionally seen in pediatric germ cell tumors (13.5% incidence), osteosarcoma (6.5%), neuroblastoma (4.4%), melanoma (3.6%), Ewing sarcoma (3.3%), rhabdomyosarcoma (1.9%), and Wilms tumor (1.3%).
Primary CNS malignancies account for about 20% of all childhood cancers, second only to leukemia. Approximately 2,200 cases are diagnosed annually in the United States in children and adolescents age 0 to 19 years, an incidence of 2.9 per 100,000 population in this age group. When nonmalignant lesions are added, the total annual incidence is about 3,750 cases. The reported annual incidence of childhood brain tumors in the United States increased by 35% between 1973 and 1994, led mainly by supratentorial low-grade gliomas and brain stem tumors diagnosed by magnetic resonance imaging (MRI), which became widely available in the mid-1980s. The reported incidence of brain tumors in infants below 12 months of age increased between 1973 and 1986 but has remained stable since then; leading histologies in infants are gliomas, medulloblastoma/primitive neuroectodermal tumor (PNET), and ependymoma.
The causes of congenital and childhood CNS tumors remain unknown. A meta-analysis from 16 international surveys revealed a male-to-female ratio of 1.29. Increased head circumference at birth is positively associated with brain cancer in childhood, with a relative risk of 1.27 per 1-cm increase after adjustment for birth weight, gestational age, and gender, suggesting brain pathology may originate in fetal development. The most common CNS tumors diagnosed in the first year of life are low-grade glioma, medulloblastoma, ependymoma, and choroid plexus tumors.
Most CNS tumors are sporadic, but some are seen in inherited disorders, especially neurocutaneous syndromes. Individuals with neurofibromatosis type 1 (NF-1) have a 10% chance of developing an intracranial tumor, including optic pathway gliomas and astrocytomas elsewhere in the brain and spinal cord. The NF-1 gene maps to chromosome 17q11.2, and loss of one NF-1 allele is evident in nearly all NF-1-associated pilocytic astrocytomas but only rarely in sporadic pilocytic astrocytomas, suggesting different mechanisms of tumor formation. In the less common neurofibromatosis type 2 (NF-2), there is a predisposition to bilateral vestibular schwannomas, with chromosome 22q deletions and NF-2 gene mutations in at least half of cases. NF-2 also predisposes to ependymomas, which commonly show a chromosome 22q deletion, but analysis of the NF-2 gene in ependymomas has revealed only a single mutation in a tumor that had lost the remaining wild-type allele.
Children with tuberous sclerosis may have a subependymal giant cell astrocytoma or ependymoma. Tuberous sclerosis genes map to chromosomes 9q and 16p, and allelic loss at these loci has been found in some subependymal giant cell astrocytomas, suggesting a tumor suppressor function. Hemangioblastomas of the cerebellum, medulla, and spinal cord are associated with von Hippel-Lindau disease, and choroid plexus tumors have occasionally been reported. Loss of chromosome 3p sequences has been described in one choroid plexus tumor, suggesting involvement of the VHL gene. There is an increased risk of brain tumors, particularly choroid plexus carcinoma, in the Li-Fraumeni syndrome, involving germ line mutations in p53 on chromosome 17. Gorlin syndrome (nevoid basal cell carcinoma syndrome), resulting from PTCH1 gene mutation, predisposes to medulloblastoma, specifically sonic hedgehog (Shh) subtype (see the following discussion).
Combined cytogenetic and molecular studies of the common sporadic childhood brain tumors have identified genomic alterations that may lead to the identification of genes contributing to tumorigenesis. Isochromosome 17q is found in half of all medulloblastomas implicating a tumor suppressor gene. Clinical outcome is now predictable by gene expression profiles. For example, overexpression of p53 in malignant gliomas of childhood is associated with adverse outcome. In medulloblastoma, low MYC oncogene mRNA expression and high TrkC neurotrophin receptor mRNA expression identify a good outcome; the desmoplastic subtype of medulloblastoma is specifically associated with the basal cell nevus syndrome (BCNS) (Gorlin syndrome), in which the molecular defect is thought to be a mutation in the human homologue of the Drosophila patched gene. Identification of a novel polymorphism in this gene allows direct detection of allelic loss in BCNS. Desmoplastic medulloblastoma in infants from affected families is reported to have a good prognosis. Conversely, the large cell, anaplastic variant of medulloblastoma and the atypical teratoid/rhabdoid tumor (ATRT) have an unfavorable prognosis. Diagnosis of these variants is based on light microscopic and immunohistochemical findings, and ATRT is further distinguished by chromosome 22q11 deletion with inactivation of the hSNF5/INI1 gene.
Environmental risk factors for CNS tumors include ionizing radiation, which in the distant past was given even for benign conditions such as tinea capitis. In children treated for acute lymphoblastic leukemia, there is a 1.39% cumulative incidence of secondary brain tumors (gliomas and meningiomas) at 20 years, and cranial irradiation is a dose-dependent predisposing factor. Cerebral PNETs may also follow cranial irradiation. Another study with 30-year follow-up found a 3.0% cumulative incidence of brain tumors (excluding meningioma) following all treatment. There is no conclusive evidence of risk from prenatal x-ray exposure, prenatal ultrasound, or from electromagnetic fields (from electric blankets or other residential exposure).
Studies have shown an association between farm and animal exposures and childhood brain tumors and a weak association with toxic exposures occurring in the household. Although not implicated in retrospective studies, maternal smoking during pregnancy was associated with slightly increased risk of brain tumors in offspring in a prospective study linking birth and cancer registries. The incidence of medulloblastoma in England declined after 1984 when multivitamin supplementation in pregnancy became widespread following reports that folate reduced the risk of neural tube defects. Maternal intake of folic acid in pregnancy was reported to be protective against PNETs, but this association could not be confirmed in a subsequent study. A seasonal variation has been noted in the incidence of medulloblastoma (but not other brain tumors) in the United States between 1995 and 2001, peaking in October. Possibly related is an observation of increased risk of medulloblastoma/PNET associated with paternal exposure to any heat source, such as electric blanket, in the 3 months prior to the index pregnancy.
PATHOBIOLOGY
Current research on CNS tumorigenesis is based on the molecular genetics of tumor-associated syndromes, rodent models of pediatric brain tumors, and technologic advances in efforts to identify genes that regulate normal and neoplastic cells in the developing CNS. Although the names of brain tumors imply that they originate from normal brain cells (e.g., astrocytoma from astrocytes), the primitive embryonal tumors of childhood that arise during brain development more likely result from aberrant regulation of neurodevelopmental genes. Recently, it has been postulated that brain tumors, like hematologic malignancies, contain small numbers of stem cells (identified as CD133-positive), which have marked capacity for proliferation and self-renewal. These stem cells can differentiate to phenotypically recognizable brain tumor cells but also maintain clonal proliferation and may be highly resistant to conventional anticancer treatment such as radiation and cytotoxic chemotherapy. Candidate stem cells have been proposed for medulloblastoma, ependymoma, and malignant gliomas. Further characterization of brain tumor stem cells may offer new insights into mechanisms of tumorigenesis and lead to novel treatment strategies based on preferential targeting of tumor stem cells.
SONIC HEDGEHOG, WNT SIGNALING, CEREBELLAR DEVELOPMENT, AND MEDULLOBLASTOMA
In the 19th century, Obersteiner described the cells of the external granular layer (EGL) in the developing cerebellum. Based on morphologic similarity, granule cell precursors (GCPs) were considered the cells of origin in medulloblastoma. Now, gene chip analysis and mouse models have linked medulloblastoma to molecular markers of GCPs. Only medulloblastomas carry these molecular markers of developing cells, whereas supratentorial PNETs do not.
The cerebellum matures through a complex interplay of genes of general CNS development and specifically cerebellar genes. For instance, Shh is a glycoprotein involved in the development of the CNS. In cerebellar development, Purkinje cells secrete Shh to activate genes in GCPs and stimulate proliferation of these cells. Activating mutations of the Shh pathway account for desmoplastic medulloblastoma in the nevoid basal cell carcinoma syndrome (Gorlin syndrome), 10% to 20% of sporadic medulloblastomas, and histologically similar tumors in mice that are heterozygous for the equivalent genes. Genome sequencing of Shh-driven medulloblastomas reveals pathway mutations involving PTCH1 (across all age groups), SUFU (infants, including cases of germ line mutation), and SMO (in adults). Functional assays in different Shh-driven medulloblastoma xenografts demonstrated that tumors harboring PTCH mutation were responsive to SMO inhibition, whereas tumors with SUFU mutation or MYCN amplification were resistant.
Wnt proteins also play a role in CNS development and have been linked to medulloblastoma. Patients with Turcot syndrome, which involves defects in the APC (adenomatous polyposis complex) gene, are susceptible to colorectal and brain tumors, notably medulloblastoma. APC is an element of the Wnt signaling pathway, and up to 30% of spontaneous medulloblastomas show mutations in APC and other components of this neurodevelopmental signaling cascade. Cell nuclear accumulation of β-catenin protein is also associated with activation of Wnt/Wg signaling, and positive immunostain for β-catenin in medulloblastoma has been associated with favorable treatment outcome even with metastatic disease at presentation.
Wnt proteins bind to receptors and modulate C-myc, a transcription factor that promotes cell cycle progression. Activation of C-myc and N-myc leads to proliferation, cell growth, and maintenance of an undifferentiated cellular phenotype; these genes are amplified in some medulloblastomas and are associated with a poor clinical outcome. The same genes are amplified by the Shh pathway. Wnt1 is required for mesencephalon/metencephalon patterning early in development. Cerebellar primordia fail to develop in mice lacking Wnt1, and mutations in other Wnt pathway components have been implicated not only in medulloblastoma but also in colon cancer, hepatocellular carcinoma, and prostate and ovarian cancers.
Deletion of chromosome 17p, the most frequently detected genetic lesion in medulloblastoma, has been identified as a cause of unrestrained hedgehog signaling. This deletion results in loss of REN(KCTD11), a novel hedgehog antagonist, thus removing a checkpoint of hedgehog-dependent events during cerebellar development and tumorigenesis. Inactivation of the tumor suppressor p53, also a resident of chromosome 17, may contribute to the growth of tumors resembling medulloblastoma in mice, but there is neither an increased incidence of medulloblastoma in humans lacking p53 nor a change in p53 within the medulloblastoma cells.
The gene Bmi1, which induces leukemia through repression of tumor suppressors, is overexpressed in many medulloblastomas and promotes cerebellar GCP proliferation. Linked overexpression of Bmi1 and patched (PTCH) in primary human medulloblastomas, together with rapid induction of Bmi1 expression on addition of Shh or overexpression of the Shh target Gli1 in cerebellar granule cell culture, implicates Bmi1 overexpression as an alternative or additive mechanism in medulloblastoma pathogenesis.
ASTROCYTES, GROWTH FACTORS, TUMOR SUPPRESSORS, AND ASTROCYTOMA
Astrocytes develop later than neurons and continue to do so long after neurogenesis stops. Astrocytes retain the capacity for division throughout life and are more susceptible to transformation. Control over astrocyte proliferation and differentiation may be divided into two main categories: cell signaling and cell cycle arrest pathways. During glial development, precursor cells respond to growth factors such as epidermal growth factor (EGF) or platelet-derived growth factor (PDGF). Their receptors (epidermal growth factor receptor [EGFR] and platelet-derived growth factor receptor [PDGFR]) are activated to stimulate tyrosine kinase activity and intracellular pathways that control proliferation and differentiation, programmed cell death, migration, cellular shape, and metabolism.
Amplification and mutation of EGFR are among the most common genetic abnormalities found in adult glioblastomas, particularly those arising de novo (as opposed to lesions progressing from fibrillary astrocytomas). These tumors contain ligands that may activate the receptor, stimulating a pathway that results in tumor progression. In addition, the receptor may be mutated to a constitutively activated form, unable to bind ligand but generating unregulated tyrosine kinase activity. Mice lacking EGFR have fewer glial fibrillary acidic protein (GFAP)-positive cells, and massive neuronal degeneration is attributed to a lack of trophic support from the missing glial cells. Amplification of EGFR is uncommon in pediatric high-grade astrocytomas, but significant differential expression of the EGFR/FKBP12/HIF-2α growth- and angiogenesis-promoting pathway has been demonstrated. High PDGFR protein expression is associated with malignant histology in pediatric glioma but is not an independent prognostic factor. Attempts to block glioma progression through disruption of the EGFR pathway have led to development of drugs targeting EGFR or PDGFR.
Ras is a downstream effector of several growth factor receptors, signaling through both mitogenic and antiapoptotic pathways. It is under control of the NF-1 gene neurofibromin, and loss of function mutations in neurofibromin underlie the NF-1 syndrome, with predisposition to optic nerve glioma, astrocytoma, and glioblastoma. Neurofibromin functions as a Ras-guanosine triphosphatase-activating protein, but it had been unclear how Ras deregulation was involved in NF-1 pathogenesis. It has now been demonstrated that neurofibromin tightly regulates the mTOR (mammalian target of rapamycin) pathway. mTOR is a highly conserved serine/threonine protein kinase that regulates many steps in cell growth and proliferation, including ribosome biogenesis and protein translation. mTOR is constitutively activated in NF1-deficient cells and tumors; aberrant mTOR activation depends on Ras and is mediated by the phosphorylation and inactivation of the tuberous sclerosis complex-2 (TSC2)-encoded protein tuberin. Tumor cell lines from NF-1 patients are highly sensitive to the mTOR inhibitor rapamycin, which has therapeutic implications.
Molecular profiling of pediatric glioblastomas identifies at least two subsets. One group is associated with Ras and Akt pathway activation and exhibits increased expression of genes related to proliferation and a neural stem cell phenotype; this subset has very poor prognosis. A second group with better prognosis is not associated with activation of Ras and Akt pathways and may originate from astroglial progenitors. Both subsets show overexpression of Y-box protein-1, which may promote oncogenesis.
The tumor suppressor phosphatase with homology to tensin (PTEN) is mutated or lost in some pediatric high-grade astrocytomas, as in primary adult glioblastoma. PTEN is expressed throughout the embryo and is critical in CNS development. PTEN mutations are thought to cause Lhermitte-Duclos disease (dysplastic gangliocytoma of the cerebellum) characterized by enlarged cerebellar foliae, distorted cerebellar cortex, abnormally myelinated axon bundles, and dysplastic neurons. Conditional inactivation of the gene in the cerebellum of developing mice has generated a mammalian model of that disease. Loss of PTEN promotes astrocytic growth and survival because it would otherwise act as a repressor of antiapoptotic signaling pathways. PTEN is also thought to regulate astrocyte migration and invasion by effects on cell shape and movement. When PTEN is overexpressed, it inhibits tumor cell movement; conversely, reduction of PTEN activity promotes cell movement, invasiveness, and perhaps metastasis. Loss of PTEN has been associated with inferior survival in childhood gliomas. PTEN is a component gene in the mTOR pathway; immunohistochemical analysis of Lhermitte-Duclos disease lesions showed high levels of phospho-AKT and phospho-S6 in the large ganglionic cells, indicating activation of the PTEN/AKT/mTOR pathway and suggesting a central role for mTOR in LDD pathogenesis, as well as potential therapeutic intervention through pharmacologic inhibition of mTOR.
The tumor suppressors retinoblastoma (Rb) protein and p53 are also regulators of cell survival and programmed cell death and are intimately involved with astrocytoma. Rb mutations in children lead to tumors in the retina, whereas mice null for Rb do not survive because of lethally abnormal patterns of neuronal development with pronounced CNS programmed cell death. Rb encodes a phosphoprotein important in cell cycle regulation, blocking transcription factors that promote proliferation. In the developing CNS, Rb is found in the ventricular zone where neuroblasts proliferate.
Rb is also involved in the regulation of apoptosis through interactions with the tumor suppressor p53, a transcription factor upregulated in response to cellular stress to facilitate arrest of the cell cycle for either DNA repair or cell death. Humans with the Li-Fraumeni syndrome lack p53 and show marked susceptibility to gliomas. p53 mutant mice develop tumors early in life.
Under conditions of Rb inactivation, many cells enter the cell cycle inappropriately and subsequently undergo apoptosis. However, Rb loss may synergize with the loss of apoptotic regulators, such as p53 to promote tumor formation. With the loss of Rb, transcription factors go unchecked, which then promotes cell proliferation. Loss of Rb might normally lead to apoptosis, but if p53 is also missing, apoptosis is impaired and tumorigenesis is enhanced. In fact, approximately 75% of adult glioblastomas show impaired apoptotic mechanisms and p53 pathway defects. Mutations in p53 were detected in one-third of childhood high-grade astrocytomas, and nearly half over-expressed the p53 protein. This finding was strongly associated with adverse outcome.
Mutations in histone and chromatin remodeling genes appear to be uniquely important in pediatric glioblastoma multiforme. Exome sequencing revealed somatic mutations in the H3.3-ATRX-DAXX chromatin remodeling pathway in nearly half of cases and recurrent mutations in H3F3A (which encodes H3.3) in one-third. H3F3A mutations were specific to GBM, highly prevalent in children and young adults, and tended to be associated with somatic TP53 mutations and alternative telomere lengthening. Increased TERC (telomerase RNA template) and hTERT mRNA (encoding telomerase catalytic component) have been associated with worse overall survival in children with non-brain stem high-grade glioma after controlling for tumor grade and extent of resection.
BRAF alterations (in particular tandem duplication and fusion BRAF-KIAA1549 on chromosome 7q34) are now widely accepted as molecular markers for pilocytic astrocytoma, in particular for the differential diagnosis of supratentorial pediatric gliomas. Besides their diagnostic use, the evidence of MAPK pathway-activating BRAF fusions and mutations in brain tumors has moreover drawn the attention of neurooncologists to the potential use of MEK1/2 and RAF inhibitors in the therapy of high- and low-grade gliomas.
Investigation of molecular aberrations in the most devastating tumor, diffuse intrinsic pontine glioma (DIPG), has lagged because most cases are diagnosed clinically without pathologic confirmation. However, high-resolution single-nucleotide polymorphism-based DNA analysis of a series of DIPGs using postmortem samples revealed recurrent changes distinct from supratentorial high-grade astrocytomas, specifically expression (and gains) of platelet-derived growth factor receptor alpha (PDGFRA) and occasional low-level gains in poly (ADP-ribose) polymerase (PARP)-1. Mutations in H3F3A (encoding histone H3.3) or HIST1H3B (encoding histone H3.1) appear to be common in DIPG. Whole-genome sequencing has identified three molecular subgroups of DIPG (H3-K27M, silent, and MYCN) and recurrent activating mutations involving the activin receptor gene ACVR1 in some cases.
TABLE 145.1 Location of Central Nervous System Tumors in Infants, Children, and Adolescents
Children with brain tumors often present with symptoms and signs of increased intracranial pressure (ICP). The ventricular or periventricular location of many tumors, and associated mass effect, results in obstruction of normal cerebrospinal fluid (CSF) pathways and ventriculomegaly. Characteristic signs are headache, vomiting, and diplopia, but the onset may be gradual and nonfocal. Fatigue, personality change, or worsening school performance may be described. Symptoms in infants are nonspecific and include irritability, anorexia, persistent vomiting, and developmental delay or regression, with macrocephaly and seemingly forced-downward deviation of the eyes (“sunset sign”). The median duration of symptoms before diagnosis was 2 months in two series; only onethird of the patients were diagnosed in the first month after onset of symptoms, but diagnostic delay is less important than tumor aggressiveness in determining survival outcome. Persistent vomiting, recurrent headache (awakening the child from sleep), neurologic findings (ataxia, head tilt, weakness, vision loss, papilledema), endocrine disturbance (growth deceleration, diabetes insipidus, precocious puberty), and stigmata of neurofibromatosis should all prompt immediate evaluation for the presence of a CNS tumor.
Symptoms and signs reflect tumor location, and in children (unlike adults), CNS tumors are divided about equally between supratentorial and infratentorial sites (Table 145.1). In a meta-analysis, the ratio of supra- to infratentorial tumors was 0.92, and infratentorial location was more common between the ages of 3 and 11 years (Table 145.2).
Supratentorial tumors, which predominate in infants and toddlers and also occur in older children, cause headache; limb weakness; sensory loss; and, occasionally, seizures, deteriorating school performance, or personality change. Seizures are a presenting symptom in 10% to 15% of children with brain tumors and may be generalized, focal motor, or psychomotor; electroencephalogram (EEG) is almost always abnormal. Most seizure-associated tumors are in the temporal lobe; frontal lobe tumors more often affect personality and movement, although seizures have been reported with frontal-parietal tumors. The incidence of underlying neoplasm in children with intractable epilepsy approaches 20%, and the increased risk of having a brain tumor is noted even 10 or more years after a diagnosis of epilepsy.
Parietal lobe tumors affect reading ability and awareness of contralateral extremities (hemineglect). Occipital lobe tumors cause visual field disturbance and occasionally, hallucinations. Suprasellar lesions may cause both visual field defects and endocrine dysfunction. The Parinaud syndrome (upgaze paresis and mild pupil dilatation with better reaction to accommodation than light, retraction or convergence nystagmus, and lid retraction) is found in pineal region tumors.
Infratentorial tumors, which predominate from ages 4 to 11 years, typically cause headache, vomiting, diplopia, and imbalance. Bilateral sixth cranial nerve palsy is a frequent sign of increased ICP. Brain stem tumors cause facial and extraocular muscle palsies, ataxia, and hemiparesis. Leptomeningeal spread occurs at diagnosis or recurrence in up to 15% of children with CNS tumors (more often in medulloblastoma/PNET) and may be asymptomatic or may cause pain, irritability, weakness, or bowel and bladder dysfunction.
TABLE 145.2 Childhood Central Nervous System Tumors Diagnosed in the United States in 2005 to 2009 by Histology and Age at Diagnosis
Data from Dolecek TA, Propp JM, Stroup NE, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro Oncol. 2012;14(suppl 5):v1-v49.
DIAGNOSIS
MRI is the preferred imaging modality for evaluation of children with symptoms suggestive of brain tumor (e.g., persistent headache of recent onset, morning vomiting) and abnormal signs on neurologic examination (head tilt, ataxia) or other clinical predictors of brain tumor (diabetes insipidus). Computed tomography (CT) may be the initial evaluation (especially if hydrocephalus or intracranial hemorrhage is suspected), but compared with CT, MRI is more sensitive (especially for nonenhancing, infiltrative tumors and leptomeningeal involvement), may generate images in any plane (e.g., axial, coronal, sagittal), and is not compromised in the posterior fossa by bone artifact. Brain tumors are typically hypo- or isointense on T1-weighted imaging and hyperintense on T2-weighted imaging. Contrast enhancement is a feature of malignant tumors, reflecting blood-brain barrier disruption. Perfusion and diffusion sequences may also be useful in assessing tumor aggressiveness. Magnetic resonance spectroscopy (MRS) can provide additional information; for example, compared to benign tumors or normal brain, malignant tumors have increased choline peaks (reflecting increased membrane turnover) and decreased N-acetylaspartate (absence of functional neurons). MRI is repeated postoperatively to confirm extent of tumor resection, although it may be difficult to distinguish tumor from surrounding edema or residual tumor from postoperative changes; for these reasons, postoperative studies should be obtained within 48 hours of surgery.
MRI of the spine has replaced myelography as the standard procedure for evaluation of spinal cord tumors, drop metastases, and leptomeningeal disease. Spine MRI is required in cases of medulloblastoma, ependymoma, choroid plexus carcinoma, ATRT, and pineal region tumors and should be obtained preoperatively if one of these diagnoses is suspected. Lumbar puncture for CSF cytology and tumor markers should also be done in children with these diagnoses. Recommended baseline and surveillance studies, based on tumor aggressiveness and patterns of recurrence, are shown in Table 145.3.
Treatment responses are determined by change in tumor size on MRI based on the most reproducible measurements from T2/fluid-attenuated inversion recovery (FLAIR)-weighted or postcontrast T1-weighted images, whichever gives the best estimate of tumor size. Measurements of the longest tumor dimension and its perpendicular should be recorded for all target lesions (up to 5) and any new lesions. In assessing response, lesions less than twice the combined thickness of the image slice and interslice gap are discounted, and cystic/necrotic elements and/or leptomeningeal disease are generally not measured. Enlargement or reduction of existing lesions should correlate with increase or decrease in two-dimensional measurement, but increased enhancement on T1 without increase in disease bulk on T2/FLAIR is not considered tumor progression, and decreased enhancement without decreased bulk on T2/FLAIR may represent altered contrast permeability rather than regression. Thus, complete or partial response (CR, PR) requires disappearance or greater than 50% reduction of all target lesions, respectively, whereas 25% increase or the appearance of new lesions or newly positive CSF cytology indicates progressive disease (PD). Imaging may be supplemented by positron emission tomography (PET), which characterizes metabolic abnormalities that distinguish high grade from low grade and residual or recurrent tumor from cerebral necrosis. Thallium-201 single-photon emission computed tomography (SPECT) is sensitive for recurrent tumors, although MRS offers similar capability and is more widely available.
TABLE 145.3 Staging and Surveillance of Children with Central Nervous System Tumors
Data from Kramer ED, Vezine LG, Packer RJ, et al. Staging and surveillance of children with central nervous system neoplasms: recommendations of the Neurology and Tumor Imaging Committees of the Children’s Cancer Group. Pediatr Neurosurg. 1994;20:254-263.
TREATMENT
SURGERY
The mainstay of therapy is surgery, which may be curative by itself in resectable congenital and benign tumors. Gross total resection is generally attempted in malignant tumors and contributes to the cure rates of high-grade astrocytoma, medulloblastoma, or ependymoma. Adoption of intraoperative MRI can improve the rate of tumor resection and may alter surgical strategy. Gross total resection (no visible tumor at completion of surgery) or radical subtotal resection (95% to 99% removal of tumor) should be confirmed by postoperative MRI. Radical resection is generally not attempted for lesions deep in the thalamus or for diffuse, intrinsic brain stem tumors; surgery is typically limited to open or closed biopsy (< 1% resection) or partial or subtotal resection (10% to 49% and 50% to 95% tumor removal, respectively). Multiple biopsies may be useful in lesions appearing heterogeneous on MRI when major resection is not attempted. The introduction of the operating microscope and ultrasonic aspirator has improved both the safety and effectiveness of surgery. Stereotactic (e.g., CT or MRI guided) techniques are used for biopsy or subtotal resection in difficult-to-reach areas such as the basal ganglia. However, even a 99% resection of a lesion that is 10 cm3 in size will leave 108 tumor cells behind, and additional postoperative treatment will be necessary to prevent the lesion from regrowing. Surgery is important for establishing a tissue diagnosis and relieving obstructive hydrocephalus. With temporary placement of an external drain or endoscopic third ventriculostomy, followed by tumor resection, it may be possible to avoid the need for a permanent ventriculoperitoneal shunt, in most patients.
Operative mortality is generally not more than 1%, but morbidity varies according to the site and extent of surgery and the condition of the child. A unique and increasingly recognized complication of midline posterior fossa tumor resection is the cerebellar mutism syndrome, consisting of diminished speech progressing to mutism, emotional lability, hypotonia, and ataxia. In two prospective trials for standard and high-risk medulloblastoma, cerebellar mutism was reported in 24% of children. In both cohorts, brain stem invasion by tumor was the only predictive feature. At 1 year from diagnosis, nonmotor speech/language deficits, neurocognitive deficits, and/or ataxia persisted in a substantial proportion of patients.
Surgery may be facilitated by intraoperative monitoring of sensory and other evoked potentials. Adjunctive measures include the use of corticosteroids, which counteract tumor edema and are often used in the perioperative period but should be tapered within 1 to 2 weeks, if possible. Patients undergoing surgery for supratentorial tumors are placed on anticonvulsants if they have had seizures or if the surgical approach is likely to cause seizures. Prophylactic anticonvulsants are generally continued for 1 week to 12 months.
RADIATION THERAPY
Radiation therapy is used for nearly all malignant CNS tumors and for some benign lesions. Based on standard x-ray (photon) therapy, doses typically administered to achieve local control range from 45 to 55 Gy, in divided fractions of 150 to 200 cGy, to the tumor as localized on MRI plus 1- to 2-cm margin. Higher doses may be given, often in smaller twice-daily doses (e.g., hyperfractionated technique), but with high total doses, there is increased risk of injuring normal brain tissue. The volume irradiated depends on tumor histology and may include an involved field or whole brain and spine. Presymptomatic craniospinal irradiation is almost always given for medulloblastoma because of its propensity to disseminate throughout the neuraxis. Doses of 36 Gy have been used conventionally, but lower doses (23.4 Gy and possibly 18 Gy) are adequate in standard-risk patients if supplemented with adjuvant chemotherapy.
Newer radiotherapy techniques may increase the effective tumor dose and limit toxicity to the surrounding brain. Three-dimensional conformal radiation therapy allows tailoring of treatment to the actual tumor volume rather than arbitrary compartments such as the whole posterior fossa. Stereotactic irradiation techniques with arc photons, in conjunction with rigid head fixation systems, permit single high-dose delivery (e.g., radiosurgery), which may be useful for management of recurrent lesions in previously irradiated patients. Intensity-modulated radiation therapy (IMRT) uses combinations of variable intensity fields from different beam directions to maximize tumor dose while minimizing dose to adjacent normal tissues. Proton therapy takes advantage of the superior physical properties of protons compared to photons, with limited penetration of particles beyond the tumor target, less tissue scatter, and little lateral dispersion. Early applications of proton therapy included ocular melanoma and skull base tumors, but the possibility of comparable tumor control with substantially decreased dose to adjacent normal tissues makes the technique potentially advantageous for many pediatric indications. Interstitial radioactive implants (brachytherapy) may be appropriate in some cases. Use of radiation sensitizers, including chemotherapy agents (carboplatin, capecitabine, tipifarnib) and gadolinium derivatives, is currently under study.
Acute side effects of radiation therapy include headache, nausea, alopecia, skin hyperpigmentation and desquamation, and a transient “somnolence syndrome,” occurring 4 to 8 weeks after treatment. Radiation therapy to large volumes is problematic in infants and toddlers because of increased neurocognitive sequelae. Dose-intensive chemotherapy may be used to defer irradiation, or involved field treatment with supplemental chemotherapy may be substituted for whole-brain irradiation (as in medulloblastoma).
CHEMOTHERAPY
Chemotherapy is used as an adjunct to radiotherapy, as primary postsurgical treatment in infants, as neoadjuvant therapy in chemotherapy-sensitive tumors (e.g., germ cell tumors), and in recurrent tumors. Effective agents include the nitrosoureas (e.g., carmustine, lomustine), vincristine, cisplatin, carboplatin, etoposide, and cyclophosphamide. Temozolomide appears promising, although response rates in phase II trials have been relatively low. Chemotherapy is generally given systemically and in combination because of complementary mechanisms of action of different agents and to subvert potential tumor resistance. The effectiveness of systemically administered chemotherapy is limited by the blood-brain barrier. Regional delivery of drugs (e.g., intra-arterial, intrathecal, intratumor) potentially circumvents this problem, and responses to intra-arterial carboplatin- or methotrexate-based chemotherapy with osmotic blood-brain barrier disruption are reported for germ cell tumors and primary CNS lymphoma. The dose intensity of conventional, systemic chemotherapy may be increased with the use of hematopoietic growth factors, which shorten the period of myelosuppression and permit the use of higher doses or shorter intervals between treatments. A related approach is the use of extremely high doses of chemotherapy, supported by autologous hematopoietic stem cell rescue, and this approach has been used for chemotherapy-sensitive recurrent tumors or as an intensive consolidation therapy for infants.
The role of adjuvant cisplatin-based chemotherapy following radiation is well established for childhood medulloblastoma, pineoblastoma, and supratentorial PNET. For standard-risk medulloblastoma, use of chemotherapy permits reduction in the dose of radiotherapy. Dose-intensive cyclophosphamide-based chemotherapy has been used following radiation therapy for standard- and high-risk medulloblastoma with the goal of reducing exposure to cisplatin. For high-grade astrocytomas, temozolomide is used routinely with and following radiation therapy, as in adults. Preirradiation chemotherapy is being tested in CNS germ cell tumors and ependymoma but has been generally ineffective in medulloblastoma/PNET and malignant gliomas, including brain stem tumors, presumably because of delaying definitive radiation therapy.
In very young patients (infants and toddlers younger than age 3 to 4 years) with malignant brain tumors, dose-intensive chemotherapy is commonly used postoperatively with the goal of deferring or avoiding radiation therapy. This approach appears effective in medulloblastoma and possibly ependymoma. Intrathecal therapy (intraventricular and intralumbar methotrexate) has also been advocated for medulloblastoma in a protocol designed to avoid radiation therapy.
Chemotherapy is commonly used for recurrent CNS tumors, and patients may receive additional irradiation as well. High-dose chemotherapy with stem cell rescue may offer effective salvage therapy for recurrent medulloblastoma at least when combined with radiation therapy in younger previously unirradiated patients. Established and newer chemotherapy agents can be tested against panels of childhood brain tumor xenografts. Doses of certain chemotherapy agents such as irinotecan may require adjustment if patients are also receiving hepatic enzyme-inducing anticonvulsant drugs such as phenytoin, phenobarbital, carbamazepine, and Mysoline.
Biologic agents in clinical trials for recurrent CNS tumors include differentiation agents (retinoic acid), agents targeting tumor growth factor receptors (erlotinib, gefitinib, imatinib, lapatinib), farnesyltransferase inhibitors (tipifarnib), agents interfering with angiogenesis (bevacizumab, cilengitide, lenalidomide), histone deacetylase inhibitors (suberoylanilide hydroxamic acid, valproic acid), heat shock protein inhibitors (17-allylaminogeldanamycin), and DNA repair enzyme inhibitors (O6-benzylguanine).
Immunotherapy would be an attractive option for treatment of pediatric CNS tumors but remains an elusive goal. Convectionenhanced delivery of immunotoxin conjugates is being evaluated in adult patients. Certain glioma-associated antigens are overexpressed on pediatric brain stem and non-brain stem gliomas and could be suitable candidates for vaccine strategies.
SPECIFIC TYPES OF PEDIATRIC BRAIN TUMORS
Pathologic diagnosis is based on the revised WHO classification published in 2007. This classification distinguishes astrocytic tumors, oligodendroglial tumors and mixed gliomas, ependymal tumors, choroid plexus tumors, and neuronal and mixed neuronal-glial tumors. Among entities newly incorporated in the revised classification are angiocentric glioma, papillary glioneuronal tumor, rosette-forming glioneuronal tumor of the fourth ventricle, papillary tumor of the pineal region, pituicytoma, and spindle cell oncocytoma of the adenohypophysis. Newly described histologic variants of pediatric tumors are pilomyxoid astrocytoma, anaplastic medulloblastoma, and medulloblastoma with extensive nodularity. In addition, the rhabdoid tumor predisposition syndrome was added to the list of familial cancer syndromes.
Some childhood brain tumors, such as supratentorial astrocytomas, are difficult to characterize in the WHO classification. A proposed pediatric classification divides tumors into glial, mixed glial-neuronal, neural, embryonal, and pineal categories. Glial tumors include astrocytoma, ependymoma, oligodendroglioma, and choroid plexus tumor. Mixed glial-neuronal tumors include ganglioglioma and subependymal giant cell tumor. Neural tumors include gangliocytoma. Embryonal tumors are PNET, ATRT, and medulloepithelioma. Pineal tumors are represented by pineocytoma.
Pathologic diagnosis is greatly facilitated by immunostaining for specific markers such as GFAP in glial tumors and neurofilament or synaptophysin in PNETs and gangliogliomas. Other markers have been useful for different tumors: epithelial membrane antigen for ependymoma, α-fetoprotein (AFP) and human chorionic gonadotropin (HCG) for germ cell tumors, S100 for neural tumors, smooth muscle actin (SMA) for ATRT, and cytokeratin for choroid plexus carcinoma (Table 145.4). An important trend in tumor pathology is the application of cytogenetic and DNA approaches. Examples are isochromosome 17q in medulloblastoma, chromosome 1p and 19q deletions in oligodendroglioma, and monosomy or deletion of chromosome 22 (hSNF/INI-1 locus) in ATRT.
CONGENITAL TUMORS
Craniopharyngiomas
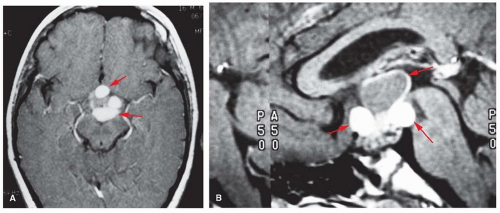
Craniopharyngiomas originate from rests of embryonic tissue located in the Rathke pouch, which later form the anterior pituitary gland. They may appear clinically at any age, even later adulthood, and constitute 6% to 10% of intracranial tumors in children. They vary from small, well-circumscribed, solid nodules to huge, multilocular cysts invading the sella turcica. The cysts are filled with turbid fluid that may contain cholesterin crystals. Craniopharyngiomas are histologically benign and may be categorized as mucoid epithelial cysts, squamous epitheliomas, or adamantinomas. Total surgical removal may be difficult because the tumor may invade the hypothalamus or third ventricle and adhere to optic nerves or blood vessels. Manifestations include short stature, hypothyroidism, and diabetes insipidus, with vision loss and signs of increased ICP. Although CT is useful for demonstrating calcification and bony expansion of the sella, MRI is preferred for better definition of the relationship of the tumor to neighboring vessels, optic chiasm and nerves, and the hypothalamus (Fig. 145.1).
A conservative approach is to drain the cyst and resect nonadherent tumor and then administer radiation therapy to the involved area. Alternatively, gross total resection may be attempted, avoiding irradiation. In addition to the standard transsphenoidal approach, there is renewed interest in the endonasal approach. Recurrence rates are similar (20% to 40%), and radical surgery is likely to be accompanied by panhypopituitarism necessitating lifelong hormonal replacement therapy, whereas irradiation has lesser hormonal sequelae but causes cognitive deficits, especially in younger children. Focused treatment by stereotactic radiosurgery (e.g., Gamma Knife) may be advantageous in this regard. Interferon α-2a and pegylated interferon α-2b have been used in recurrent craniopharyngiomas.
Epidermoid Cysts
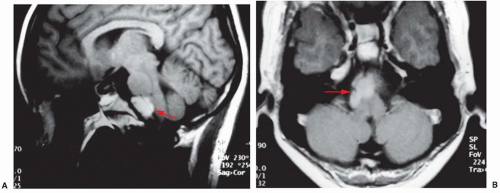
Epidermoid cysts (cholesteatomas) account for about 2% of all intracranial tumors. They arise within skull tables or adjacent to the dura usually in the suprasellar region, skull base, brain stem or cerebellopontine angle, or within a ventricle. They arise from epithelial cells retained during neural tube closure, have a pearly appearance, and may contain cyst fluid with cholesterol crystals. Clinical onset is usually in young adulthood, with hearing loss, hemifacial spasm, or trigeminal neuralgia. MRI demonstrates a lesion with low T1- and high T2-weighted signal intensity (Fig. 145.2); involvement of multiple cranial nerves and the internal carotid artery is common. Treatment is surgical resection with radical removal of the tumor capsule. Dermoid tumors are also cystic, and sebaceous gland secretions impart a dark yellow color to the cyst fluid. Intracranial dermoids are associated with Goldenhar syndrome (i.e., oculo-auriculo-vertebral dysplasia). Treatment is surgical resection. Epidermoid and dermoid cysts occasionally undergo malignant transformation into squamous cell carcinoma. Arachnoid cysts are CSF collections within arachnoid membranes, often in the sylvian fissure, presenting with headache and occasionally seizures. Diffusion-weighted imaging can be helpful in distinguishing arachnoid cysts from epidermoid cysts. Surgery is indicated for arachnoid cysts, which are symptomatic (obstructive hydrocephalus, seizures); options include cystectomy, fenestration, or cyst-peritoneal shunting.
TABLE 145.4 Immunohistochemical, Cytogenetic, and Molecular Characteristics of Childhood Brain Tumors
Tumor
Immunohistochemistry
Cytogenetics
Molecular Pathology
Pilocytic astrocytoma
GFAP
+7q34
BRAF fusion
NF1 mutation
KRAS mutation
MAP kinase activation
High-grade astrocytoma
GFAP
+1q
10q23 LOH
TP53 mutation
MAP kinase activation
IDH1/2 mutation
EGFR mutation
Medulloblastoma
Synaptophysin
NSE
β-Catenin (nuclear)
Iso(17)
-6
PTCH mutation
TP53 mutation
APC mutation
MYC amplification
MYCN amplification
FOXG1 upregulation
sPNET
Synaptophysin
S100
NSE
-9p21
-14q
p16 downregulation
Ependymoma
GFAP
EMA
+1q25
-6q25.3
CDKN2A/B mutation
EGFR amplification
ATRT
SMA
EMA
Vimentin
INI-1 negative
del(22q11)
SMARCB1mutation
Choroid plexus Ca
Pancytokeratin
Vimentin
E-cadherin
TP53 mutation (germ line)
Germ cell tumors
AFP
β-HCG
Iso(12p)
XXY
P14 mutation
c-kit mutation
Oligodendroglioma
S-100
Del(1p/19q)
IDH1/2 mutation
FUBP1 mutation
Meningioma
Vimentin
EMA
-22q
NF2 mutation
PDGFR-β activation
GFAP, glial fibrillary acidic protein; LOH, loss of heterozygosity; NSE, neuron-specific enolase; sPNET, supratentorial primitive neuroectodermal tumor; EMA, epithelial membrane antigen; ATRT, atypical teratoid/rhabdoid tumor; SMA, smooth muscle actin; Ca, carcinoma; AFP, α-fetoprotein; β-HCG, β-human chorionic gonadotropin.
Data from Pfister S, Hartmann C, Korshunov A. Histology and molecular pathology of pediatric brain tumors. J Child Neurol. 2009;24(11):1375-1386.
FIGURE 145.1 Axial (A) and sagittal (B) MRIs of a craniopharyngioma.
FIGURE 145.2 Sagittal (A) and axial (B) MRIs of a pontomedullary epidermoid cyst.
Teratomas
Teratomas occur in infants and young children. Prenatal diagnosis of an intracranial teratoma has been reported. Mature teratomas are generally lobulated and cystic, often containing differentiated tissues such as bone, cartilage, teeth, hair, or intestine. Immature and malignant teratomas are less common; posterior fossa immature teratoma in an infant with trisomy 21 has been reported. Teratomas tend to occur in the pineal region, presenting with Parinaud syndrome and hydrocephalus. They comprise about 4% of childhood intracranial tumors and also occur in the spine. Treatment is surgical resection.
Chordomas
Chordomas develop from remnants of the embryonic notochord. Half are in the sacrococcygeal region, one-third at the sphenoid-occipital junction, and the remainder elsewhere along the spinal column. They are rare, accounting for less than 1% of CNS tumors, and usually remain asymptomatic until adulthood. These tumors are locally invasive and cause vision loss or cranial nerve dysfunction. They may invade the nasopharynx or intracranial sinuses and may extend into the neck causing torticollis.
Chordomas have a smooth, nodular surface that resembles cartilage; the characteristic histologic feature is the presence of large masses of physaliferous cells, round or polygonal cells arranged in cords and having large cytoplasmic vacuoles containing mucin. Chordomas grow slowly but may recur after excision. A malignant variant is distinguished by mitotic spindle cells and may metastasize to the lung. Chordoma should be suspected in a patient presenting with multiple cranial nerve palsies or erosion of the skull base. Treatment is surgical resection, although gross total resection is rarely achieved, occasionally with postoperative irradiation. Radiation therapy may be used postoperatively or at recurrence. Proton radiotherapy is advocated because it may confer effective local control and possibly better cosmetic outcome than with conventional irradiation.
Choroid Plexus Tumors
Choroid plexus tumors are rare congenital tumors arising in the choroid plexus of the lateral or fourth ventricles. They may cause symptoms shortly after birth, and most are diagnosed before age 2 years. The first manifestation is likely to be macrocephaly resulting from hydrocephalus, with bulging fontanelle and split sutures noted in infants. Excess CSF production arising from the tumor may approach 2,000 mL/day. The majority are papillomas (WHO grade I), hamartomatous lesions which grow into the ventricle and tend not to be invasive. Papillomas are often calcified on CT scan and enhance with contrast; they must be distinguished in the infant age group from ependymomas, which lack calcification. Atypical papillomas (WHO grade II) have a higher growth fraction (mitotic rate by MIB-1 >2%). Total excision of localized lesions may be curative and stops the excessive CSF production, but malignant transformation of incompletely resected papillomas has been reported.
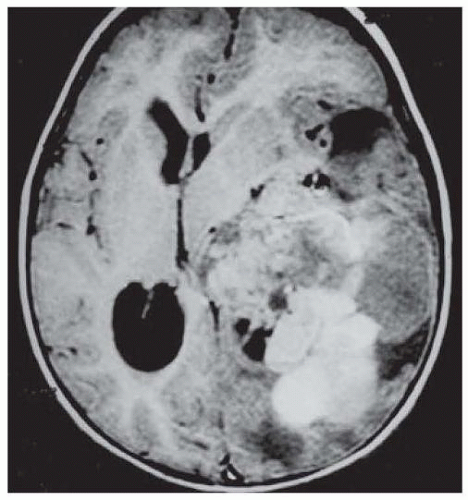
Choroid plexus carcinomas (WHO grade III) typically invade the brain parenchyma and cause hydrocephalus (Fig. 145.3). Carcinomas are more heterogeneous than papillomas on imaging due to focal necrosis and surrounding vasogenic edema; in nearly half of cases, there is disseminate in the leptomeninges. Choroid plexus carcinomas may be a manifestation of Li-Fraumeni syndrome, and evidence should be sought for germ line mutation in p53. Adjuvant chemotherapy or irradiation has been advocated for carcinomas, even following apparent gross total resection, owing to their invasiveness and guarded prognosis (median survival 6 months in one series). For patients undergoing subtotal resection of carcinomas, both chemotherapy and craniospinal irradiation are recommended.
FIGURE 145.3 MRI of a choroid plexus carcinoma of the lateral ventricle with parenchymal invasion.
Colloid Cysts
Colloid cysts presumably arise from the anlage of the paraphysis. These lesions grow in the anterior superior portion of the third ventricle as small, white cysts filled with homogeneous, gelatinous material. They do not usually cause symptoms until adulthood when there may be intermittent hydrocephalus. Disturbance of the limbic system may cause emotional and behavioral changes in some instances. Colloid cysts are T2 hyperintense on MRI without enhancement. Treatment is surgical excision. Endoscopic procedures are gaining acceptance as an alternative to microsurgical resection.
Gangliogliomas
Gangliogliomas are rare, glioneural tumors that account for 2.5% of pediatric brain tumors. The peak age incidence is 10 to 20 years. They arise mainly in the temporal lobe but also in other lobes or spinal cord. The usual syndrome is a long history of refractory epilepsy. Imaging reveals a solid to cystic lesion with calcification on CT and hypometabolic activity on PET. Total surgical resection is recommended, and prognosis for long-term seizure control is excellent. Malignant transformation to glioblastoma may follow incomplete resection. Radiation therapy and chemotherapy are of uncertain benefit.
Desmoplastic infantile ganglioglioma (DIG) is a rare cerebral glioneural tumor of early childhood. DIGs are large hemispheric tumors that are generally benign, but deep lesions may be aggressive. Treatment is resection. Related glioneural tumors include desmoplastic astrocytoma of infancy (DACI) and pleomorphic xanthoastrocytoma (PXA), which usually cause seizures and headache.
Dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) is a rare, benign entity characterized by loss of normal cerebellar cortical architecture and may be associated with macrocephaly and mental retardation. MRI appearance is a wellcircumscribed lesion with characteristic laminar pattern of alternating high and low T2 signal. The disorder has been linked with Cowden disease, with increased risk of breast, uterine, and thyroid cancer and harbors abnormalities in the PTEN/AKT pathway, consistent with germ line mutation in the PTEN tumor suppressor gene in Cowden disease.
Dysembryoplastic Neuroepithelial Tumor
Dysembryoplastic neuroepithelial tumor (DNT) is a rare, low-grade (WHO grade I) mixed neuronal and glial tumor often associated with intractable seizures in children and young adults. DNTs comprise up to 17% of underlying diagnoses in epilepsy surgery series. Cortical dysplasia is found in 30% of cases. Enhancement on MRI is patchy and multifocal; perfusion-weighted MRI and MRS may aid in diagnosis. Unlike other epilepsy-associated glioneuronal tumors, where seizures arise within the lesion, seizures associated with DNT are considered to arise from the surrounding dysplastic brain; thus, extended resection is preferred over lesionectomy alone. Risk factors for recurrent seizures include age older than 10 years and epilepsy duration more than 2 years prior to diagnosis.
ASTROCYTOMAS
Low-Grade Astrocytomas
Low-grade astrocytomas (WHO grades I and II) include superficial lesions in the cerebral or cerebellar cortex and deeper infiltrative lesions of the optic pathways and midbrain. Cerebellar astrocytomas constitute about one-third of posterior fossa tumors. The peak incidence is early in the second decade. These tumors arise most often in the lateral cerebellar hemispheres rather than in the vermis. Symptoms of clumsiness and unsteadiness may be present for months and eventually are accompanied by intermittent morning headache and vomiting. Midline lesions have a shorter symptom interval. Truncal ataxia, dysmetria, and papilledema are usually present at diagnosis. Head tilt may be present, but other cranial nerve palsies or long-tract signs are infrequent and suggest brain stem invasion. Cerebellar astrocytomas may be primarily cystic or solid, and cystic lesions may have a nodule (Fig. 145.4). The typical juvenile pilocytic astrocytoma (WHO grade I) contains areas of compact fibrillated cells with microcysts and eosinophilic structures called Rosenthal fibers. These lesions have greater than 90% survival with surgery alone. The tumor and associated cyst should be removed completely, and if residual tumor is identified postoperatively, reresection should be considered, as irradiation will not delay tumor progression. Diffuse astrocytomas (WHO grade II) may infiltrate the brain stem and be more difficult to resect entirely, with more guarded prognosis for survival.
Only gold members can continue reading. Log In or Register to continue