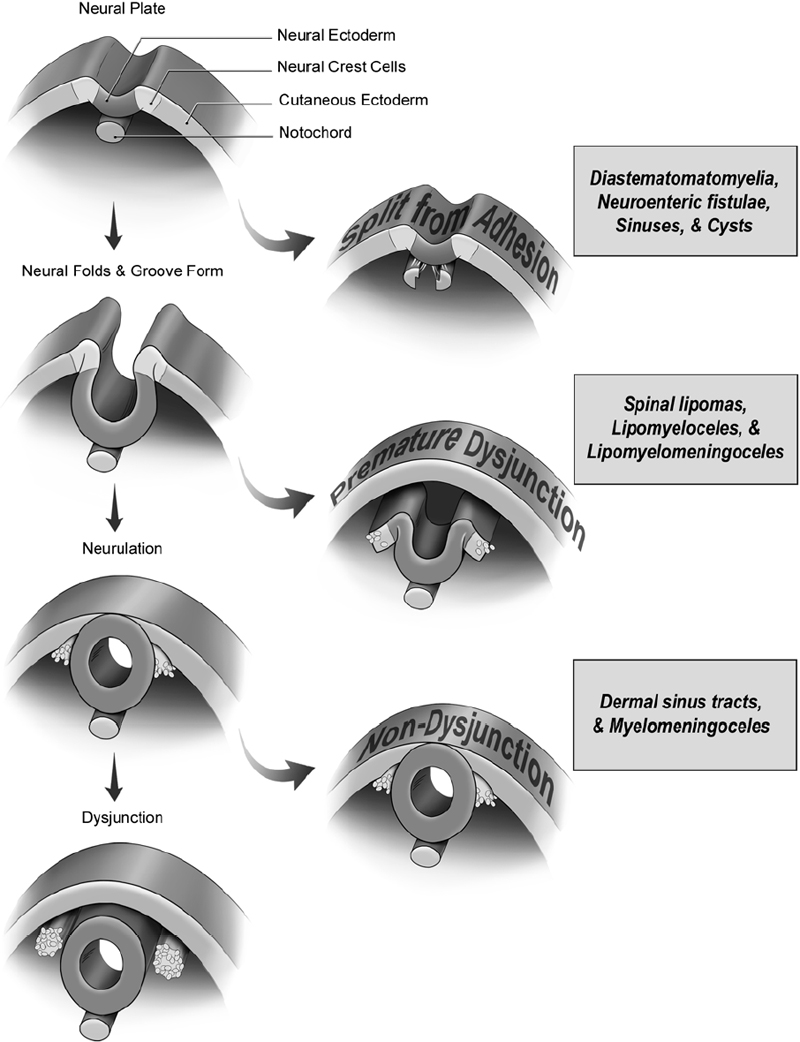
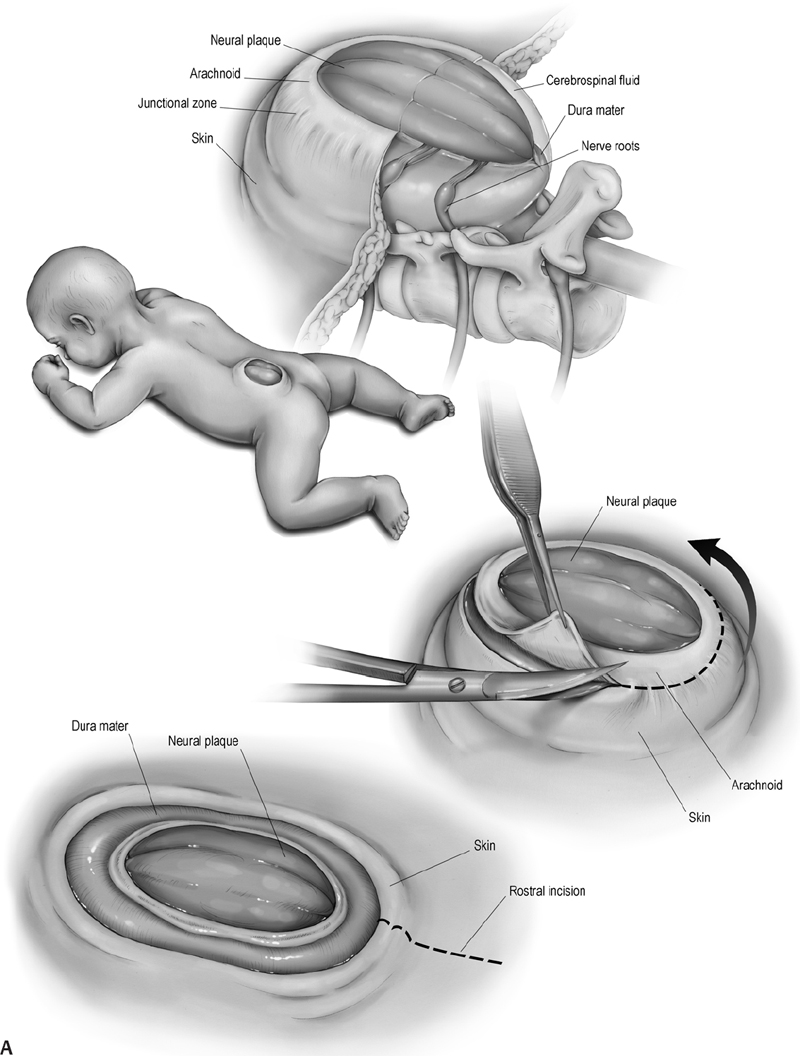
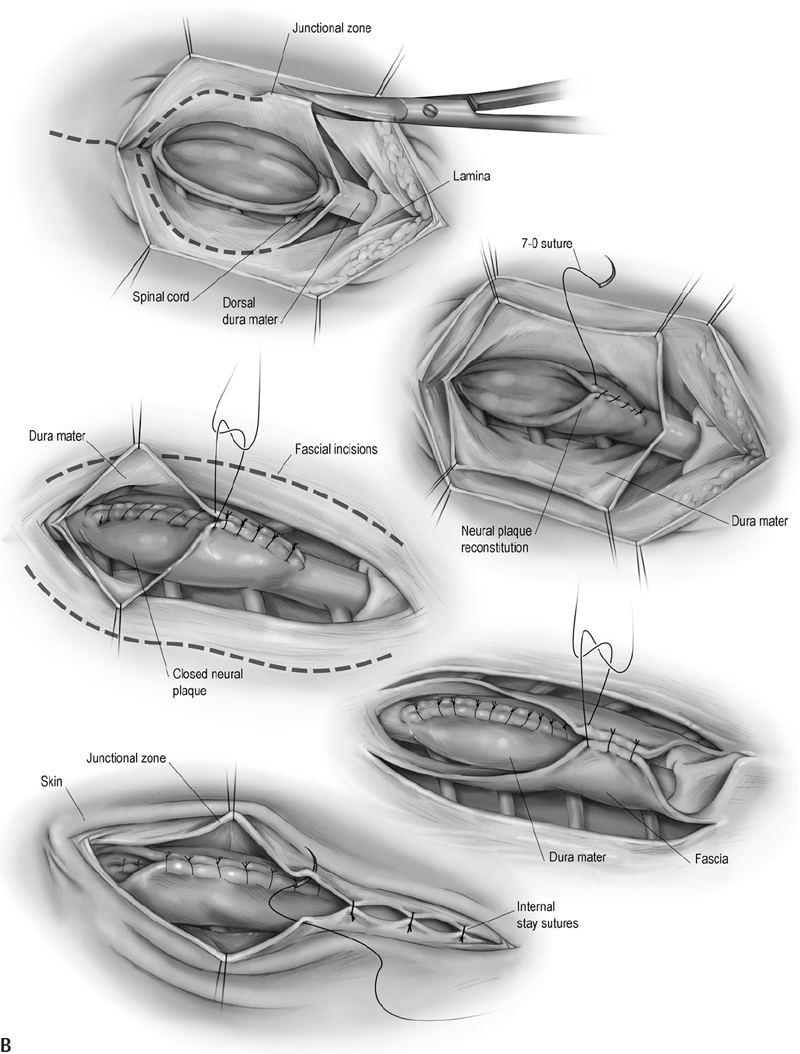
16 Congenital Anomalies I. Key Points – Most developmental disorders of the spine occur in either the upper cervical or lower thoracic and lumbar regions due to defective spinal cord embryogenesis and vertebral column formation (Fig. 16.1). In the general workup, screen with ultrasound for antenatal diagnosis and use sagittal magnetic resonance imaging (MRI) for spinal dysraphism. Employ computed tomography (CT) or CT myelogram to better delineate bony malformations and hydrocephalus. Use renal ultrasound to rule out pelvic kidney, unilateral agenesis, and horseshoe kidney. Fig. 16.1 Defects during different stages of spinal cord embryology lead to congenital anomalies of the spine. II. Anomalies of Notochord Formation Diastematomyelia – Background • The spinal cord is divided vertically into two hemicords, each with its own central canal, surrounding pia, and set of anterior and posterior nerve roots. • May result from bifurcation of the developing notochord around an adhesion between the endoderm and ectoderm. The split notochord may influence the formation of two neural tubes and subsequent hemicords and vertebral formation. Consequently, it is common to have associated bony anomalies (spurs) at the site of diastematomyelia. • Accounts for 5% of congenital scoliosis and roughly 30 to 40% of myelomeningoceles – Signs, symptoms, and physical exam • Most individuals have nonspecific symptoms and tethered cords requiring surgical attention. • The overlying skin in 66% of cases shows nevi, hypertrichosis, lipomas, dimples, hemangiomas, dermal sinus tracts, or dermoids. – Neuroimaging • The conus is low lying in 75%, often with an associated thickened or fatty filum.1 • The hemicords usually reunite below the cleft, enveloped in a single dura, with no spur or fibrous band found in 60% of cases (Pang Type II).2 In contrast, the presence of two dural tubes is always associated with a fibrous or bony spur within the cleft in the remaining 40% (Pang Type I). • The type of spur, bony (50 to 60%) or fibrous (40 to 50%), is likely determined by the amount of trapped mesenchyme between the hemicords. – Treatment • Untether the cord, coagulate and divide tethering fibrous bands extending from the dura to the spinal cord, and excise any bony spurs. – Surgical pearls • Remove the spur before untethering the cord to prevent retraction of the cord against the bony septum. • Remove dural cuff and arachnoid covering the bony spur to prevent regeneration of spur. Split Notochord Syndrome – Background • Deviation or splitting of the notochord secondary to a retained link between the endoderm and ectoderm. The most severe form, a dorsal enteric fistula, is a communication between the bowel and the dorsal skin. Dorsal enteric fistulae traverse the prevertebral soft tissues, vertebral bodies, the spinal canal, and its posterior elements. Any portion of the tract may involute or fibrose, leaving fistulae or cysts. • Dorsal enteric sinuses open on the skin surface • Dorsal enteric cysts are found in the intraspinal or paraspinal compartments. – Signs, symptoms, and physical exam • Dorsal enteric fistula present in newborn with a bowel ostium on the back. • Intraspinal enteric cysts usually present between 20 and 40 years of age as episodic local or radicular pain that may progress to myelopathy. – Workup • Cyst incision, drainage, and pathologic examination to rule out infectious processes – Neuroimaging • Radiography will help determine the presence and degree of vertebral clefting. • Follow with CT/MR imaging to define the degree of fistulation or cyst involvement. – Treatment • Complete surgical excision is the best treatment. Chemical arachnoiditis resulting from cystic material often produces dense adhesions that make later operations more difficult. III. Anomalies of Dysjunction Spinal Lipomas3 – Background • Fat and connective tissue masses attached to the spinal cord and meninges • Lipomyelomeningoceles and lipomyeloceles (84%) and intradural lipomas (4%) may arise from premature separation of the neuroectoderm from the cutaneous ectoderm, providing space for mesenchymal tissue to invade the canal of the neural tube and promote fat formation. However, the exact cell lineage and subsequent pattern of differentiation to adipocytes have not yet been undetermined. Fibrolipomas of the filum terminale (12%) probably result from an abnormality of retrogressive differentiation. – Signs, symptoms, and physical exam • Multiple cutaneous anomalies including midline/paraspinal mass, focal hirsutism, dermal sinus, rudimentary tail, atretic meningocoele, and/or capillary hemangioma. Neurologic deficits affect 60 to 70% of patients, but younger children more often have a cutaneous sign caused by tethering or by the lipoma mass. • Pain, an unusual finding in infants, is often the most common presenting symptom in older children and adults. It often becomes worse with activity and is rarely radicular in nature. • Lipomyelomeningoceles and lipomyeloceles • Intradural lipomas may present with an ascending monoparesis or paraparesis, spasticity, cutaneous sensory loss, or defective deep sensation. – Neuroimaging • Use MRI for surgical planning and to detect associated malformations (split cord, arachnoid cysts, meningoceles, and syringomyelia). T1-weighted MRI helps visualize lipomas and T2-weighted MRI is useful for meningoceles and syringomyelia. A low-lying conus is found in many patients and suggests tethering in those with small filum lipomas. • CT myelography is less informative and more invasive but may be used if MR is contraindicated. CT may also be utilized to assess for bony anomalies. – Treatment • Surgically debulk/resect lipomatous mass and untether if symptomatic, or do so when the conus medullaris is low lying in conjunction with a terminal filum lipoma.4 • Asymptomatic cases: there is debate over conservative treatment versus prophylactic surgery. • For subdural lipomas, the goal is decompression, not detethering or complete excision. • If considerable lipoma is found within the central canal, debulk to reduce pressure on neural tissue but avoid full resection, for which a myelotomy may be needed. – Surgical pearls • When the neural placode is positioned dorsal to the open spinal canal, consider the use of an ultrasonic aspirator or laser to carefully debulk the lipoma. Dorsal Dermal Sinuses5 – Background • Thin, squamous, epithelia-lined channels associated with (epi)dermoid tumors • Due to a focal incomplete separation of neuroectoderm from cutaneous ectoderm – Signs, symptoms, and physical exam • History of recurrent meningitis despite antibiotics due to bacterial passage along the tract or from chemical irritation if the cyst (e.g., cholesterol crystals) ruptures • A patent sinus tract may leak cerebrospinal fluid (CSF). • A hairy nevus or hyperpigmented skin may be seen. – Neuroimaging • T1- or T2-weighted MRI usually reveals the dermal sinus tract running at an oblique angle through the underlying tissues, and diffusion MRI can define its margins. • Anomalies of the bone may range from absent to focal or multilevel spina bifida. – Treatment • Treat early with excision of the dimple, tract, and any intradural connections or masses. Myelocele and Myelomeningocele6 – Background • A failure of closure of the neural tube linked to folate deficiency in pregnancy. The neural and cutaneous areas of ectoderm remain contiguous, forcing mesenchymal tissue that normally forms the posterior elements to become displaced laterally. – Signs, symptoms, and physical exam • Newborn with an exposed, raw, red tissue placode positioned in the midline • Level determines severity of neurologic deficits, with the most common being lumbosacral or thoracolumbar. Hydrocephalus is common secondary to Chiari II malformation. – Workup • Maternal serum a-fetoprotein (MSAFP) levels may be used to screen at 16 to 18 weeks gestation. Use amniocentesis if MSAFP and ultrasound suggest an abnormality. – Neuroimaging • Associated anomalies include syringohydromyelia, diastematomyelia, lipoma, arachnoid cyst, dermoid/epidermoid, and vertebral anomalies (e.g., hemivertebrae). • Chiari II observed to varying degree in all patients with myelomeningoceles. • The spine is rarely imaged prior to closure/repair, whereas postoperative neurologic deterioration requires imaging. Symptomatic retethering is a diagnosis of exclusion. If the spine is normal, image the brain to rule out hydrocephalus or shunt malfunction. – Treatment • Surgically close within 48 hours to stabilize neurologic deficits and prevent infection • Manage postoperative complications: CSF leak, retethering, and/or hydrocephalus – Surgical pearls (Fig. 16.2) • Reconstruct the neural tube and close the pia, dura, thoracolumbar fascia, and skin to prevent meningitis and to protect functional tissue in the neural placode IV. Anomalies of Caudal Cell Mass Tight Filum Terminale Syndrome – Background • Incomplete retrogressive differentiation is the suspected etiology. – Signs, symptoms, and physical exam • Possibilities are bladder dysfunction, sensory changes, orthopedic deformities (e.g., clubfoot), radiculopathy, and scoliosis associated with a short, thick filum and low-lying conus. – Neuroimaging • Utilize T1-weighted MRI. Roughly half of patients have a thick, fibrotic filum with tethered cord syndrome, and 23% have small fibrolipomas. The conus is usually low lying, and 25% have a small hydromyelia within the conus that resolves after untethering. – Treatment • Resect the filum when clinical criteria for tethered cord syndrome are satisfied. – Surgical pearls • Ensure that there are no nerve roots adherent to the undersurface of the filum. Fig. 16.2 (A) The multilayer closure begins with an initial circumferential incision following the arachnoid-skin junction to isolate the neural plaque and spinal cord. The skin incision is continued rostrally in the midline to observe the caudal-most intact lamina and the dura beneath it. (B) An inferior and lateral dissection toward the caudal edge of the defect isolates the dura. The caudal defect is extended and the residual dura dissected. In the midline, the dura is then approximated following that of the lateral arachnoid. The lumbosacral fascia is identified and incised bilaterally with dissection from the posterior iliac crest and sacrospinalis muscle, with care taken to not disrupt the sacral fascial attachments. The lateral edges of the fascial flaps are then folded toward the midline and sutured in place over the dorsal surface of the dura. The subcutaneous tissue, if present, is closed. The placement of sutures in the area where the dura joins the dermis will provide for tight internal sutures that will reduce tension during later skin closure. Fibrolipomas of the Filum Terminale – Background • Fibrolipomas result from aberrations of filum terminale development. • A long and fibrous filum projects from the conus and penetrates through the subarachnoid space and dura, connecting dorsally to the first coccygeal segment. – Signs, symptoms, and physical exam • Often asymptomatic, patients may not present with symptoms until late adulthood. • Following presentation, monitor patient for signs of tethering. – Neuroimaging • May be found in conjunction with a tight filum terminale – Treatment • Resect lipomas of the filum to untether the spinal cord to prevent progressive orthopedic deformity and neurologic deficit and to preserve sphincter function Syndrome of Caudal Regression – Background • Likely caused by a disruption of the caudal mesoderm before the fourth week of gestation, resulting in an abnormal distal spinal cord and vertebrae • Approximately one-sixth of patients have diabetic mothers. – Signs, symptoms, and physical exam • Spectrum includes lower extremity fusion (sirenomelia), lumbosacral agenesis, anal atresia, malformed external genitalia, bilateral renal aplasia, and pulmonary hypoplasia with Potter facies. The vast majority of patients present with a neurogenic bladder. – Neuroimaging • The spinal canal is very stenotic above the last intact vertebra. • MRI may demonstrate a wedge-shaped cord terminus, which is characteristic. • Lipomas or a lipomyelomeningocele may cause tethering. – Treatment • A large proportion of patients have a tethered cord that may require untethering. V. Anomalies of Segmentation Klippel-Feil Syndrome7 – Background • Congenital fusion of two or more cervical vertebrae. Spectrum runs from vertebral body fusion (congenital block vertebrae) to fusion of the entire vertebrae. • Due to a failure, during 3 to 8 weeks of gestation, of normal segmentation of prospective somatic mesoderm into discrete cervical somites. This failure may involve disordered notch signaling and the PAX gene family. • Type I: fusions at C1 with or without associated caudal fusions, most often with other congenital anomalies. Type II: fusions no higher than C2-C3. Type III: fusions caudal to C2-C3. Type IV: synonymous with Wildervanck syndrome. – Signs, symptoms, and physical exam • Often asymptomatic, although the classic triad (occurring in less than 50%) involves a short neck, low posterior hairline, and limited neck motion best elicited with flexion-extension or lateral bending. C1-C2 fusions often present with pain in childhood, whereas lower cervical fusions may present in the second or third decade when symptomatic junctional degeneration occurs. • May occur in association with basilar impression or atlantooccipital fusion • Associated with scoliosis, facial asymmetry, torticollis or neck webbing, Sprengel deformity (congenital elevation of the scapula), synkinesia, or cervical ribs • Systemic congenital anomalies: deafness (in 30%), unilateral renal agenesis, cardiopulmonary (ventricular septal defect most common), and a variety of central nervous system findings • Symptoms rarely relate directly to vertebral fusion, but may result from adjacent hypermobile nonfused segments leading to degenerative arthritic changes or instability. – Workup • Electrocardiogam, chest x-ray, and renal ultrasound. Audiology testing may contribute to workup. – Neuroimaging • Initial studies to visualize fusion should include anteroposterior, lateral, and open-mouth odontoid views followed by serial lateral flexion-extension C-spine radiography to evaluate for instability of the atlanto-occipital, atlantoaxial, and subaxial joints. • Image the thoracic and lumbar spine to rule out scoliosis and other abnormalities. • Flexion-extension MRI is indicated if preliminary studies suggest instability. – Treatment • Consider activity modification, bracing, and/or traction to reduce symptoms, delay surgery, and prevent major neurologic deficits that may occur, even after minor trauma. • Operate in case of progressive symptomatic segmental instability or neurologic deficits. – Surgical pearls • At the risk of further limitations in mobility, occasional fusion of adjacent unstable nonfused segments may be needed. • Sublaminar wires should be used with caution in cases of posterior instrumented fusion because they carry an unacceptably high risk of neurologic injury and may not be applicable in children with anomalous vertebrae. Common Clinical Questions 1. Myelomeningoceles result from: A. An anomoly of notochord formation. B. Premature dysjunction. C. Nondysjunction. D. Regression of the caudal cell mass. 2. The majority of spinal lipomas result from: A. An anomoly of notochord formation. B. Premature dysjunction. C. Nondysjunction. D. Regression of the caudal cell mass. 3. A tight filum terminale results from: A. An anomoly of notochord formation. B. Premature dysjunction. C. Nondysjunction. D. Regression of the caudal cell mass.
 Typically present before 6 months of age with bladder dysfunction common in 50% and half presenting without neurologic compromise
Typically present before 6 months of age with bladder dysfunction common in 50% and half presenting without neurologic compromise
 Neuroorthopedic syndrome (33 to 50%): lower-extremity deformities such as clubfoot, length discrepancies, scoliosis, trophic ulcers, and hip subluxations
Neuroorthopedic syndrome (33 to 50%): lower-extremity deformities such as clubfoot, length discrepancies, scoliosis, trophic ulcers, and hip subluxations
 No significant difference in risk of neurological deterioration
No significant difference in risk of neurological deterioration
 The posterior neuropore may remain open too long, decompressing the ventricular system and allowing the posterior fossa to close when premature and small.
The posterior neuropore may remain open too long, decompressing the ventricular system and allowing the posterior fossa to close when premature and small.
 Hydrocephalus usually results within 48 hours of myelomeningocele closure.
Hydrocephalus usually results within 48 hours of myelomeningocele closure.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


