19 Tumors of the Spine
I. Key Points
– Spine tumors are broadly organized into three general categories depending on the spine compartment invaded by the neoplasm: extradural, intradural extramedullary, and intradural intramedullary (Fig. 19.1).
– Overall, magnetic resonance imaging (MRI) is the best modality for viewing neoplasms in and around the spine and thus best for determining the localizing compartment of a neoplasm.
– Determining which compartment is affected is essential to every aspect of managing the patient. The crucial information yielded includes the primary differential diagnosis (Fig. 19.1), the pathophysiologic mechanism connecting the presence of the tumor to the clinical syndrome, and the appropriate surgical strategy.
II. Metastatic Epidural Spine Tumors
– Background
• Metastatic spine tumors are more prevalent than primary spine tumors.1,2
• Out of 1.5 million annually incident cancer cases, 10% result in symptomatic secondary metastases, and the bony spine is the third most common site of metastases. The thoracic spine is the most commonly afflicted.1
• Peak incidence: fourth to sixth decade. Men are more likely to be afflicted.1
• The two most likely origins of spinal metastasis are breast and lung cancer.1
• The two most common mechanisms of spread to the spine are hematogenous spread and direct extension.1
– Signs, symptoms, and physical exam
• Presentation is a factor of systemic tumor spread, amount of bony destruction, extent of neural compression, and tumor growth rate.1,3
• Physical examination is crucial to elicit appropriate signs of neurologic dysfunction, pain, and palpable masses. A thorough history is essential to elicit risk factors (e.g., cigarette smoking).
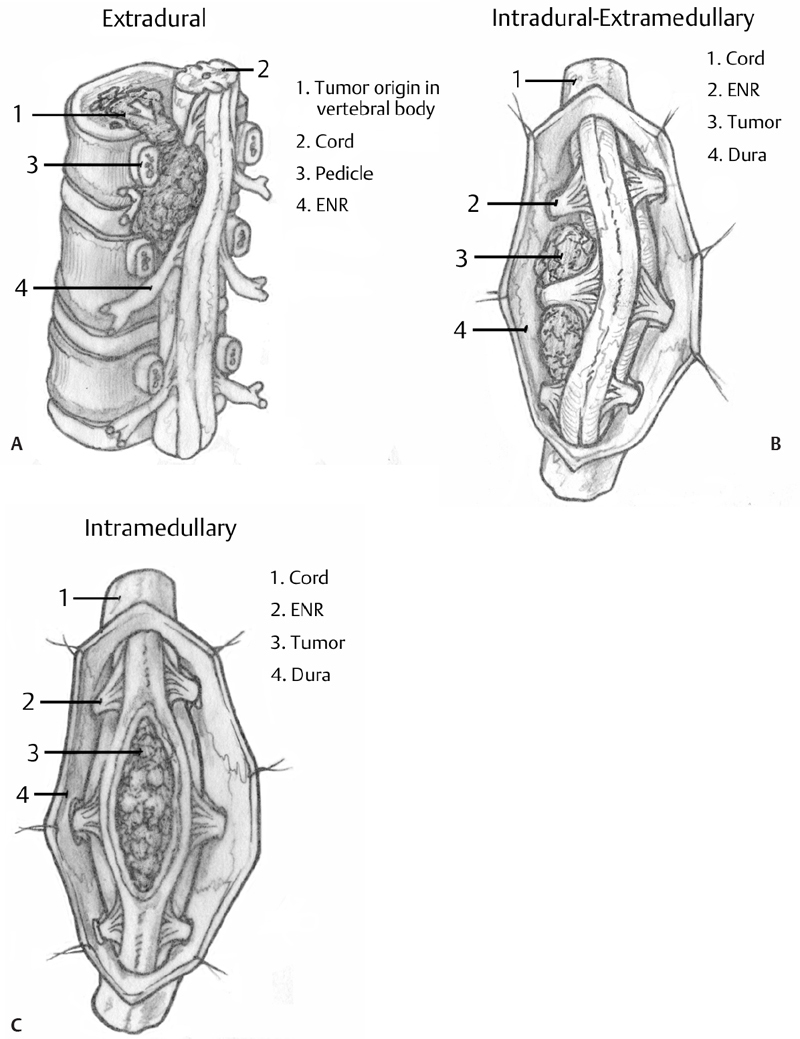
Fig. 19.1 (A–C) Artistic rendition demonstrating the structural relationship of differently compartmentalized neoplasms to the spinal cord and adjacent structures. Differential diagnosis based on compartmentalization of the neoplasm (from Khanna AJ, ed. Magnetic Resonance Imaging for Orthopaedic Surgeons. Thieme; Pg. 318, Fig. 12.1; pg. 329, Fig. 12.17; pg. 332, Fig 12.22).
• Pain is the most common initial complaint and can be categorized as radicular (radicular compression or foraminal stenosis), mechanical (spinal instability due to compromised vertebral bodies and adjacent structures), or local pain.
• Motor and autonomic dysfunctions are the second most common signs of metastatic epidural spinal cord compression (MESCC).1
• Sensory dysfunction signs such as anesthesia, hyperesthesia, and paresthesias are the next most common. When signs are of myelopathic origin, patients describe symptoms as being distributed in a bandlike fashion.
• Compromise of bowel or bladder function and loss of the ability to ambulate are crucial prognosticators.
• Other important signs include those of systemic neoplastic disease, such as marked weight loss.
– Workup and neuroimaging
• Diagnostic blood work should include prostate-specific antigen assays, blood chemistry, and blood cell counts.
• The gold standard for imaging of metastatic spine disease is MRI without and with contrast because of the quality of visualization of the bone—soft tissue interface, elucidating compression or invasion of osseous, neural, and paraspinal structures. T2-weighted images and T1 contrast-enhanced images have the highest yield.1,3
• Computed tomography (CT) is useful for detailed rendition of osseous anatomy, myelography studies (in cases of MRI contraindication), evaluation of vascular supply, and whole body scans to detect the tumor of origin.
• Plain radiographs are relatively insensitive but can be useful to screen for pathologic fractures, spinal deformities, sclerotic lesions, lytic lesions, and large masses.
– Treatment
• For the most part, treatment goals are therapeutic, as curative treatment is possible only in select cases (i.e., solitary renal cell carcinoma). Therapeutic goals include preserving neurologic function, mechanical stabilization, and pain relief.
• Surgical candidacy factors include functional status (the most prognostic factor of postoperative neurologic function),1 age, and life expectancy (with 3 months considered a minimum). Objective scales are available for patient selection.3
• Surgical goals should be achieving optimal resection, decompression, and stabilization (i.e., vertebral body reconstruction and pedicle instrumentation).
• Nonsurgical candidates can be managed via minimally invasive means such as vertebroplasty or kyphoplasty.
• Adjuvant treatments include pharmacotherapy (tumoricidal and palliative) as well as radiation therapy (conventional radiotherapy or stereotactic radiosurgery).1
– Surgical pearls
• Although surgical exposure is always of utmost importance, these patients often have poor healing capabilities due to systemic cancer spread, previous use of corticosteroids, and radiation exposure to the surgical field. For this reason, surgical exposure and wound closure should be done with plastic surgery as needed to avoid postoperative wound complications.
III. Primary Epidural Spine Tumors
– Background
• Ten percent of the tumors that affect the bony spine are primary epidural spine tumors, and they occur more frequently in men than in women.2
• The most frequently encountered primary spine tumors are chordomas, chondrosarcomas, osteosarcomas, and Ewing sarcoma.2,4 Chordomas are slow-growing tumors and represent 1% of primary spine tumors.5 Chondrosarcomas are responsible for 7 to 12% of primary spine tumors.2 Osteosarcomas are less common but are the most common malignant tumor of osseous origin. Predisposing factors include adolescence, a family history of retinoblastoma, and previous exposure to ionizing radiation.2
• Among children, eosinophilic granulomas and Ewing sarcoma are the most common benign and malignant tumors, respectively.2
• Hemangiomas are the most common benign and plasmacytomas the most common malignant primary epidural spine tumors in adults.2
• Other, rare tumors include giant cell tumors, aneurysmal bone cysts, osteoid osteomas, and osteoblastomas.2,4
– Signs, symptoms, and physical exam
• Chordomas: The most common symptoms are back and neck pain. Nearly a third of patients will demonstrate signs of neurologic deficit, and physical exam may identify a palpable mass.2,5
• Chondrosarcomas: Signs and symptoms of radiculopathy, myelopathy, cauda equina syndrome, and nocturnally exacerbated focal pain are common.2,4
• Osteosarcomas: The most common presentation is an insidious development of back pain that is exacerbated nocturnally.2,4
• Ewing sarcoma: Patients most commonly present with symptoms of pain and local inflammation, and this is frequently misdiagnosed as infection. Signs and symptoms of systemic illness such as weight loss and fever are also common.2,4
• Plasmacytomas: In addition to symptoms of pain and neurologic deficit, patients commonly present with diffuse osteoporosis, bone fractures, and osteolytic bone lesions.2,4
– Workup and neuroimaging
• Histopathological analysis of the lesion is crucial for selecting appropriate intervention in respect to each tumor origin (i.e., prognosis and sensitivity to pharmacotherapy or radiotherapy).3
• MRI is the gold standard for chordomas and chondrosarcomas, which are both hyperintense under T2 weighting.2,5 However, chondrosarcomas can be differentiated from chordomas and other tumors via gadolinium enhancement, which yields a characteristic ring-and-arc pattern.4
• The gold standard for imaging osteosarcomas is positron emission tomography (PET) scanning due to its ability to measure bone turnover. Highly mineralized tumors are hyperintense on T1-weighted MRI. Tumors with low mineralization are hyperintense on T2-weighted imaging.2
• Ewing sarcoma lesions can be detected via conventional radiography because they have a mottled, moth-eaten appearance. The possibility of distant metastases should be ruled out via a whole-body CT scan.2
• In case of suspicion of multiple myeloma, blood cell counts, blood chemistries, and serum/urine electrophoresis should be obtained. These studies may show evidence of renal failure, infections, hypercalcemia, anemia, or Bence-Jones proteins. Diagnostic bone marrow biopsies are also appropriate. The lesions can be imaged via CT or MRI (T2-weighted) and are usually not hot (i.e., no increased uptake) on a bone scan.2,4
– Treatment of primary epidural spine tumors
• The most important prognostic factors for primary spine tumor include tumor identity, location or spread, size, and histologic grade.
• Ideally, the neoplasms should be completely excised via en bloc resection with wide surgical margins and avoidance of tumor breach. Breach is correlated with a high local recurrence rate.2
• Local recurrence of the neoplasm can be avoided with concomitant adjuvant therapy such as chemotherapy and radiotherapy (note, however, that osteosarcomas and chondrosarcomas are radioresistant).
• Ewing sarcoma can be managed with chemotherapy, in particular a combination of four drugs: doxorubicin, cyclophosphamide, vincristine, and dactinomycin. In addition, Ewing sarcoma is radiosensitive and can be treated via conventional radiotherapy.2
• Plasmacytomas do not need surgical intervention. They can be managed with pharmacotherapy (i.e., chemotherapy and adjuvant bisphosphonates) and radiotherapy. Surgical intervention is reserved for cases of marked spinal instability.2
– Surgical pearls
• Primary epidural tumors: En bloc spondylectomy is the gold standard for radio-insensitive tumors (chordoma, chondrosarcoma, etc.). Such techniques should be attempted by surgeons experienced in such procedures, often in conjunction with thoracic surgery, orthopedic surgery, general surgery, vascular surgery, and plastic surgery.
IV. Intradural Extramedullary Spinal Cord Tumors
– Background
• Intradural extramedullary tumors are the second most common tumor type in the spine.6
• Primary lesions in this compartment arise from perineural coverings of nerve roots or from the meninges; thus meningiomas, schwannomas, neurofibromas, and paragangliomas account for the majority of tumors in this compartment.6
• Although the majority of tumors are benign, they can result in significant neurologic dysfunction.
– Signs, symptoms, and physical exam
• Symptomatology is usually of insidious onset. The most common complaint is localized or radicular (and thus not pathognomonic) pain. Other signs and symptoms include gait problems, weakness, paresthesia, impotence, and autonomic dysfunction.6
• The presence of an acute headache should raise suspicion of subarachnoid hemorrhage.
• Physical examination findings include Brown-Sequard syndrome and signs of long tract involvement, such as Babinski sign, clonus, and hyperreflexia.
• Extramedullary tumors can sometimes be distinguished from intramedullary tumors by the fact that intramedullary tumors spare dorsal tracts whereas extramedullary tumors normally affect all sensory modalities.6
– Workup and neuroimaging
• Imaging is essential to identifying the tumor compartment (i.e., intramedullary versus extramedullary).
• MRI is the gold standard, but when it is contraindicated, CT myelography is the imaging of choice.
• Meningiomas, schwannomas, neurofibromas, and paragangliomas are all hyperintense on MRI T2-weighted images. They are all iso- or hypointense in T1-weighted images.6
• Schwannomas can be distinguished from meningiomas because schwannomas may demonstrate cystic changes within the tumor and appear as focal areas of increased signal in T2-weighted images. In contrast, meningiomas rarely develop cystic changes. Schwannomas are also often dumbbell shaped.6
• Paragangliomas demonstrate strong enhancement upon administration of gadolinium contrast.
• Metastatic lesions should be suspected in a patient with a previous history of cancer, and in such a case imaging should include a whole-body scan to assess systemic spread of the metastatic neoplasm.1
– Treatment
• Complete microsurgical excision of intradural extramedullary tumors is optimal but not always possible due to factors that dictate surgical approach (i.e., anterior or posterior cord location) and the degree to which neural structures are involved by the neoplasm. For example, neurofibromas normally grow from the central root as an enlargement of the nerve itself, making complete surgical resection very challenging. This is in contrast to schwannomas, which typically involve only one fascicle of a nerve root, making it possible to completely dissect the mass and preserve the remainder of the nerve root function.6
• When complete resection is not possible without neurologic compromise, partial resection that avoids neurologic compromise is at times appropriate. This decision is individualized to patient, and factors to consider are patient age, neurologic status at presentation, tumor histopathology and size, and factors predisposing to local recurrence, such as a medical history of neurofibromatosis.
• The risk of local recurrence can be reduced postoperatively with adjuvant chemotherapy and radiotherapy for sensitive tumors.
• Radiosurgery can be considered in cases of recurrence, multiple lesions, and an absence of compressive myelopathy.
– Surgical pearls
• Careful attention should be given to removing such tumors from the spinal cord rather than attempting to initially dissect the tumor—spinal cord interface. Often this requires initial internal tumor debulking. In this way, the spinal cord is not manipulated in its most compressed state.
V. Intramedullary Spinal Cord Tumors
– Background
• Intramedullary spinal cord tumors make up roughly 6 to 8% of all tumors of the central nervous system.6
• The two most common intramedullary spinal cord tumors are low- or high-grade astrocytomas and ependymomas.6
• Low-grade astrocytomas are more common in children whereas ependymomas are more prevalent in adults.6
• High-grade astrocytomas often have a poor prognosis because they are highly infiltrative and have a high rate of recurrence.6
– Signs, symptoms, and physical exam
• Intramedullary spine tumors have a nonspecific presentation. Initial signs and symptoms can be of insidious onset or follow a trivial injury.
• Presenting signs and symptoms include radicular pain, localized pain, dysesthesia, paresthesia, spasticity, torticollis, extremity weakness, Brown-Sequard syndrome, and autonomic dysfunction.6
• In children, these tumors can present as a failure to achieve developmental milestones.6
• Intramedullary tumors that localize to the cervical spine may also be accompanied by hydrocephalus.2
– Workup and neuroimaging
• MRI is the gold standard for imaging intramedullary spinal cord tumors.
• T1-weighted images reveal the solid tumor component when performed before and after administration of gadolinium contrast.
• T2-weighted images allow visualization of cystic elements and the cerebrospinal fluid.
• When viewed axially, astrocytomas are located eccentrically in the spinal cord and may display heterogeneous enhancement under T1 weighting. In contrast, ependymomas are visualized most often in the center of the spinal cord when imaged axially. In addition, ependymomas most often exhibit homogeneous enhancement.6
• Plain radiographs may be useful preoperatively as a baseline reference for managing spinal deformity, as in the case of scoliosis patients.
• A CT myelography study is acceptable when MRI is contraindicated.
– Treatment
• Histologic grade and preoperative neurologic function are the most significant prognostic factors for surgical management of intramedullary spinal cord tumors. High-grade astrocytomas have approximately an 80% mortality rate within the first 6 months of diagnoses. Conversely, it is possible to completely surgically resect and cure ependymomas.6
• The approach to surgical resection varies with tumor type and tumor characteristics.
• Astrocytomas have a grayish yellow, glassy appearance. They should be resected beginning at the midpoint of the neoplasm and moving in an inside-to-outside fashion. The tumor should be debulked until the border between the spinal cord and tumor can be reasonably demarcated.6
• Ependymomas have a red or dark gray appearance and display a characteristic visible boundary in relation to the spinal cord. Ependymomas can be resected en bloc and separation at the tumor–spinal cord boundary can be achieved by applying a microsurgical laser or plated bayonet in the axial direction.
• Electrophysiologic monitoring can be used to assess the patient’s neurologic status throughout the procedure. This includes somatosensory evoked potentials (SSEPs) and motor evoked potentials (MEPs).6
– Surgical pearls
• Often these tumors can be easily suctioned off of the normal parenchyma of the spinal cord when they are necrotic (highgrade astrocytoma) or when a good plane is noted between the tumor and cord (ependymoma or low-grade astrocytoma). Extreme care should be taken to avoid dissecting the plane between tumor and cord with instruments as these maneuvers contuse and stretch the cord; rather, suction should be used for debulking and sharp dissection in locations where the tumor is focally tethered to the cord.
Common Clinical Questions
1. The least common histology of metastatic tumor arising in the spine is:
A. Breast adenocarcinoma
B. Prostate adenocarcinoma
C. Non–small cell lung adenocarcinoma
D. Transitional cell carcinoma of the bladder
2. Classic initial presentations of metastatic epidural spinal cord compression include all of the following except:
A. Abdominal pain
B. Bladder retention
C. Localized back pain
D. Gait imbalance
3. The most important predictor of survival in patients with spine tumor is:
A. Presence of metastases in the liver
B. Histopathology of the lesion
C. Number of spinal levels involved
D. Local invasion into paraspinal tissues
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


