Chapter 28 Congenital Anomalies of the Skull
Introduction
Congenital anomalies of the skull can arise any time during gestation and must be distinguished from anomalies that arise after birth. During the first 4–6 weeks of development from conception, neural crest cells in the fetal head region migrate and differentiate into mesenchymal cells that form the bones of the face, while the cranial base and base of the skull are derived from the occipital somitomeres. Within the flat cranial bones, the mesenchyme differentiates directly into bone through membranous ossification. The neurocranium is the portion surrounding the brain, and it develops directly from mesenchyme derived from the occipital somitomeres. The viscerocranium is derived from neural crest and it forms the cartilaginous bones of the face [Sadler and Langman, 2009]. The neurocranium is divided into the membranous part, forming the flat bones of the cranial vault, and the chondrocranium, forming the cartilaginous bones of the base of the skull. The chondrocranium develops by fusion of a number of cartilaginous structures, which ossify by endochondral ossification to form the base of the skull. Posteriorly, the base of the occipital bone is formed from parachordal cartilage and three occipital sclerotomes. Anteriorly, the sphenoid and ethmoid bones are formed from the hypophyseal cartilages and trabeculae cranii. On either side of the medial plate, the ala orbicularis and ala temporalis form the sphenoid bones, and the periotic capsule forms the temporal bones. The membranous neurocranium (dura mater) ossifies to form the cranial vault through bone spicules, which progressively radiate from primary ossification centers near the center of each bony plate toward the periphery, where the sutures develop. Membranous bones enlarge during fetal and postnatal life by the apposition of new layers to the outer surface of the skull (ectocranial bone deposition), while endocranial osteoclastic bone resorption occurs on the inner surface. Growth of the cranial bones is directly related to brain growth, and premature fusion of cranial sutures can be related to the cessation of brain growth (e.g., primary microcephaly).
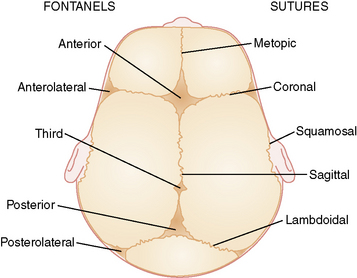
There are six fibrous areas where two or more cranial bones meet (fontanels). The five major sutures are the metopic, sagittal, coronal, squamosal, and lambdoid sutures; the six fontanels are the anterior (1), anteriolateral (sphenoidal) (2), posterolateral (2), and posterior (1) fontanels (Figure 28-1). The presence of the sutures and fontanels allows the bones of the skull to overlap each other (termed molding) during the birth process. Different sutures become ossified at different times, with the metopic suture being the first to ossify at 4–7 months and the remaining sutures not completely ossifying until adulthood.
Craniosynostosis versus Deformational Plagiocephaly
The term craniostenosis (literally “cranial narrowing”) is used to describe an abnormal head shape that results from premature fusion of one or more sutures, while craniosynostosis is the process of premature sutural fusion that results in craniostenosis. In clinical usage, the term craniosynostosis is used more widely, perhaps in an effort to distinguish deformational nonsynostotic head shapes from those caused by underlying sutural synostosis, but the two terms are often used interchangeably. Plagiocephaly is a nonspecific term used to describe an asymmetric head shape, which can result from either craniosynostosis or cranial deformation, and differentiation between these two causes is critical to determining the proper mode of treatment (i.e., surgery vs. physical or molding techniques). Craniosynostosis is usually treated with a neurosurgical procedure involving partial calvariectomy, while deformational plagiocephaly usually responds to early physical therapy, repositioning, and/or cranial orthotic therapy if those measures are unsuccessful [Graham and Smith, 2007; Graham et al., 2005].
In an otherwise normal fetus, prenatal relaxation of normal growth-stretch tensile forces in the underlying dura across a suture for a significant period of time during late fetal life can result in craniosynostosis [Graham et al., 1979, 1980; Higginbottom et al., 1980; Graham and Smith, 1980; Koskinen-Moffett et al., 1982]. This may also occur when the lack of growth stretch is caused by a deficit in brain growth, as in severe primary microcephaly. Experimental prolongation of gestation in pregnant mice, resulting in fetal crowding after installation of a cervical clip, has been shown to result in craniosynostosis [Koskinen-Moffett and Moffet, 1989]. The degree of craniosynostosis was greatest among those fetuses located more proximally in the uterine horns, where the crowding was most severe. The most common cause of craniosynostosis in otherwise normal infants is constraint of the fetal head in utero [Graham et al., 1979, 1980; Higginbottom et al., 1980; Graham and Smith, 1980; Koskinen-Moffett et al., 1982; Koskinen-Moffett and Moffet, 1989; Sanchez-Lara et al., 2010]. When external fetal head constraint limits growth stretch parallel to a cranial suture, it may lead to craniosynostosis of an intervening suture between the constraining points. Sagittal craniosynostosis (the most common type of craniosynostosis) usually is isolated and occurs in an otherwise normal child. The constrained suture tends to develop a bony ridge, especially at the point of maximal constraint between the biparietal eminences. Such ridging can easily be palpated or visualized on skull radiographs, and three-dimensional cranial computed tomography (3D-CT) allows the ridge to be seen even more clearly (Figure 28-2).
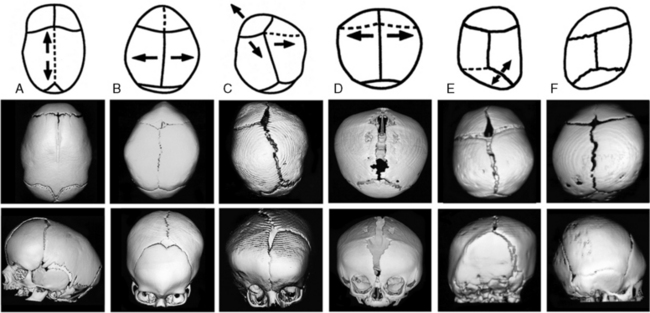
Craniosynostosis is usually recognized shortly after birth from the abnormal shape of the head and lack of molding resolving to normal. Early closure of a fontanel, head asymmetry, and/or palpable ridging along a closed suture can be presenting features. With synostosis, cranial radiographs may reveal sclerosis of the suture with no apparent intervening sutural ligament, but it may be difficult to distinguish an overlapping suture from synostosis. On cross-sectional images, dense ridging over the suture may be evident, particularly with sagittal and metopic synostosis. If there is uncertainty as to whether sutures are truly synostotic, 3D-CT can provide a more accurate appraisal (see Figure 28-2).
In general, craniosynostosis begins at one point and then spreads along a suture [Koskinen-Moffett and Moffet, 1989; Cohen and MacLean, 2000]. At the fused location, there is complete sutural obliteration, with nonlamellar bone extending completely across the sutural space, while further away from the initial site of fusion, the sutural margins are closely approximated with ossifying connective tissue. As age increases, there is a tendency for more of the suture to become synostotic, with synostosis usually beginning at only one location in most cases [Koskinen-Moffett and Moffet, 1989]. Synostosis prevents future expansion at that site, and the rapidly growing brain then distorts the calvarium into an aberrant shape, depending upon which sutures have become synostotic (see Figure 28-2). The earlier the synostosis takes place, the greater the effect on skull shape. Craniosynostosis may be caused by many different mechanisms, such as mutant genes, chromosome disorders, storage disorders, hyperthyroidism, or failure of normal brain growth The entire topic of craniosynostosis has been comprehensively reviewed by Cohen [Cohen and MacLean, 2000].
Sutural Anatomy and Head Shape
Different terms have been used to describe the different head shape alterations caused by craniosynostosis, with the resultant head shape dependent on the suture involved. A long, keel-shaped skull with prominent forehead and occiput is termed dolichocephaly or scaphocephaly. This head shape is usually associated with premature sagittal suture closure and a palpable ridge toward the posterior end of the suture (see Figure 28-2A). Sagittal synostosis must be distinguished from deformation of the infant cranium due to persistently sleeping on the side of the head (more common in prematurely born infants) or breech-head deformation sequence. Individuals with scaphocephaly and dolichocephaly have a decreased cephalic index (CI) of less than 76 percent (CI = head width/head length × 100 percent).
Premature fusion of both coronal sutures produces a high, wide forehead with a short skull, resulting in brachycephaly (see Figure 28-2), while fusion of one coronal suture produces an asymmetric head shape termed plagiocephaly. When coronal craniosynostosis occurs, it is important to examine the patient carefully for associated anomalies that might suggest a recognizable genetic syndrome. Evaluation of the limbs, ears, and cardiovascular system is quite helpful in diagnosing syndromes associated with coronal craniosynostosis. Limb defects, such as syndactyly, brachydactyly, carpal coalition, or broad, deviated thumbs and/or halluces, can suggest an associated genetic syndrome and indicate what types of molecular analysis to pursue. It is also important to examine both parents for similar anomalies, carpal coalition, and/or facial asymmetry, since these findings may represent variable expression of an altered gene in a parent.
Premature fusion of both the coronal and sagittal sutures generally leads to a tall, tower-like skull (turricephaly), with more severe synostosis of multiple sutures producing a tall, pointed skull (acrocephaly or oxycephaly). In this condition, the limitations of calvarial expansion are so extreme that there may be limited room for brain growth (Figure 28-3a). Synostosis of multiple cranial sutures is more likely to result in elevated intracranial pressure and to require shunting for hydrocephalus. In extreme cases, a cloverleaf head shape can result from multiple suture synostosis, usually with signs of increased intracranial pressure and a “beaten copper” radiographic appearance of the inner table of the skull (see Figure 28-3b). There may also be optic atrophy, proptosis, and loss of vision. Combinations of sutural synostosis, such as sagittal plus coronal, are also referred to as compound craniosynostosis, and multiple suture synostosis usually has a genetic basis.
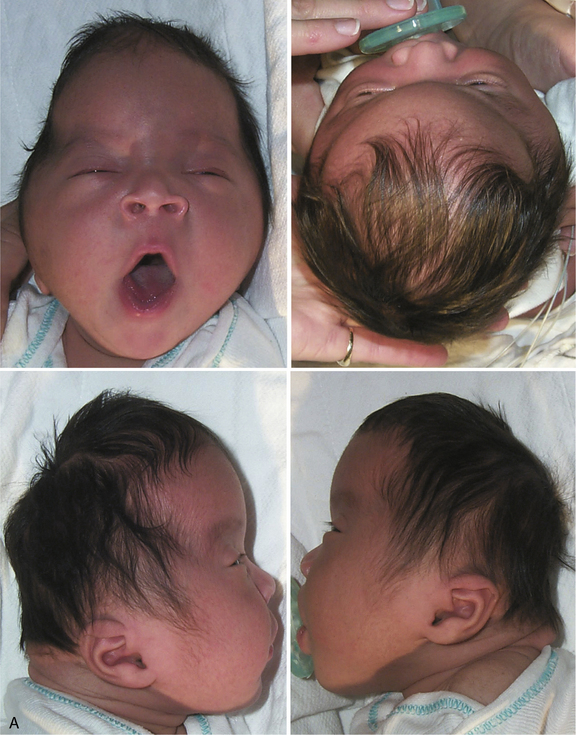
A triangle-shaped skull (trigonocephaly) is caused by premature fusion of the metopic suture (see Figure 28-2). The similarity in epidemiological features between sagittal and metopic craniosynostosis suggests that prenatal lateral constraint of the frontal part of the head may be a frequent cause of metopic craniosynostosis. Examples of constraint-induced metopic synostosis have included a monozygotic triplet whose forehead was wedged between the buttocks of her two co-triplets, and an infant whose head was compressed within one horn of his mother’s bicornuate uterus [Graham and Smith, 1980]. Syndromic metopic synostosis can also occur, and trigonocephaly is seen in a variety of syndromes, some of which are associated with mental retardation or chromosome anomalies.
Unilateral lambdoid synostosis results in trapezoidal plagiocephaly, which differs from deformational posterior plagiocephaly due to supine positioning and torticollis, and from synostotic anterior plagiocephaly due to unicoronal synostosis (see Figure 28-2). Unlike coronal synostosis, facial structures and orbits are usually not affected by lambdoid synostosis. Radiographic signs include trapezoidal cranial asymmetry, small posterior fossa, and sutural sclerosis with ridging; however, sole reliance on skull radiographs and clinical signs can lead to misdiagnosis, so it is best to confirm the diagnosis of suspected lambdoid synostosis with a 3D-CT scan, which clearly images the involved suture(s) and permits secure diagnosis. Among 232 patients referred for either deformational posterior plagiocephaly or craniosynostosis, only 4 patients (3.1 percent) manifested clinical, imaging, and operative features of true unilambdoidal craniosynostosis [Huang et al., 1996]. These features included a thick bony ridge over the fused suture, with contralateral parietal and frontal bulging, and an ipsilateral occipitomastoid bulge, leading to tilting of the ipsilateral skull base and a downward/posterior displacement of the ear on the synostotic side. In contrast, infants with deformational, nonsynostotic posterior plagiocephaly had a parallelogram-shaped head, with forward displacement of the ear and frontal bossing on the side ipsilateral to the occipitoparietal flattening, accompanied by contralateral occipital bossing [Graham et al., 2005].
Plagiocephaly (which translates literally from the Greek term plagio kephale as “oblique head”) is a term used to describe asymmetry of the head shape, when viewed from the top [Graham and Smith, 2007]. The term deformational plagiocephaly should suffice to distinguish this type of defect and its proper type of management. The side of the plagiocephaly is usually indicated by the bone that has been most flattened by the deforming forces (usually the occiput for infants who sleep on their backs). Deformational plagiocephaly is usually not associated with premature closure of a cranial suture, but since craniosynostosis can also be caused by fetal head constraint, when both deformational plagiocephaly and craniosynostosis occur together, the diagnosis can be difficult, requiring complex management.
Epidemiology of Craniosynostosis
The incidence of craniosynostosis is 3.4 per 10,000 births, and it is usually an isolated, sporadic anomaly in an otherwise normal child. About 8 percent of all craniosynostosis cases are familial. Familial types of craniosynostosis are most frequent in coronal synostosis, accounting for 14.4 percent of coronal synostosis, 6 percent of sagittal synostosis, and 5.6 percent of metopic synostosis [Lajeunie et al., 1995, 1996, 1998], while lambdoidal synostosis is almost never familial. The frequency of associated twinning is increased, and most twin pairs are discordant, especially with sagittal and metopic synostosis, which would tend to support fetal crowding as a cause for these types of synostosis; concordance for coronal synostosis is much higher for monozygotic twins than for dizygotic twins, suggesting that many cases of coronal synostosis have a genetic basis [Lajeunie et al., 1995].
Sagittal synostosis is the most common type of craniosynostosis, accounting for 50–60 percent of cases and occurring in 1.9 per 10,000 births, with a 3.5:1 male:female sex ratio [Lajeunie et al., 1996]. Only 6 percent of cases are familial, with 72 percent of cases sporadic and no paternal or maternal age effects noted. Twinning occurred in 4.8 percent of 366 cases, with only one monozygotic twin pair being concordant [Lajeunie et al., 1996].
Coronal craniosynostosis is the second most frequent type of craniosynostosis (accounting for 20–30 percent of cases). Unilateral coronal craniosynostosis can be either genetic or due to fetal head constraint from an aberrant fetal lie, multiple gestation, or small uterine cavity [Graham et al., 1980; Higginbottom et al., 1980]. Approximately 71 percent of unilateral coronal craniosynostosis is right-sided, and 67 percent of vertex presentations are in the left occiput transverse position, possibly explaining the prevalence of right-sided, unilateral, coronal craniostenosis [Graham et al., 1980]. Nonsyndromic coronal craniosynostosis occurs 0.94 per 10,000 births, with 61 percent of cases sporadic, and 14.4 percent of 180 pedigrees familial. Bilateral cases occur much more frequently than unilateral cases, and coronal synostosis is more frequent in females (male to female ratio 1:2). The paternal age is statistically older than average (32.7 years), and these data have been interpreted as being consistent with fresh dominant mutation and autosomal-dominant inheritance with 60 percent penetrance, when the synostosis has a genetic basis [Lajeunie et al., 1995].
Metopic synostosis occurs in about 0.67 per 10,000 births, making it the third most frequent type of craniosynostosis (accounting for 10–20 percent of patients). Like sagittal synostosis, metopic synostosis is more frequent in males (3.3:1 male: female ratio) and seldom familial (5.6 percent of cases) [Lajeunie et al., 1998]. There is no maternal or paternal age effect, and the frequency of associated twinning was 7.8 percent of 179 pedigrees studied, with only 2 twin monozygotic pairs concordant [Lajeunie et al., 1998].
Familial craniosynostosis is usually transmitted as an autosomal-dominant trait with incomplete penetrance and variable expressivity. A wide variety of chromosomal anomalies have also been associated with craniosynostosis. This emphasizes the importance of chromosomal analysis for patients with syndromic craniosynostosis in whom a recognizable monogenic syndrome is not apparent, particularly when there is associated developmental delay and growth deficiency. As comparative genomic hybridization becomes more widely utilized, this will be even more useful than standard cytogenetics for many such cases. In addition, craniosynostosis can also occur as a component of numerous syndromes, many of which manifest phenotypic overlap and genetic heterogeneity [Cohen and MacLean, 2000] Craniosynostosis syndromes with a demonstrated mutational basis include Apert’s syndrome, Crouzon’s syndrome, Pfeiffer’s syndrome, Saethre–Chotzen syndrome, Jackson–Weiss syndrome, Boston craniosynostosis, Beare–Stevenson cutis gyrata syndrome, and fibroblast growth factor receptor 3 (FGFR3)-associated coronal synostosis; however, the efficiency of actually detecting a mutation for a given syndrome varies from about 60 percent for Crouzon’s syndrome to 98 percent for Apert’s syndrome [Cohen and MacLean, 2000].
Secondary craniosynostosis can occur with certain primary metabolic disorders (e.g., hyperthyroidism, rickets), storage disorders (e.g., mucopolysaccharidosis), hematological disorders (thalassemia, sickle cell anemia, polycythemia vera, congenital hemolytic icterus), brain malformations (e.g., holoprosencephaly, microcephaly, encephalocele, or overshunted hydrocephalus), and selected teratogenic exposures (e.g., diphenylhydantoin, retinoic acid, valproic acid, aminopterin, fluconazole, cyclophosphamide) [Cohen and MacLean, 2000].
Inability to demonstrate a mutation does not rule out a genetic basis for the craniosynostosis, and not every person with a pathogenic mutation manifests craniosynostosis. Bilateral coronal synostosis often lacks sutural ridging and usually has a genetic pathogenesis, suggesting that all such patients should be screened for mutations. Among 57 patients with bilateral coronal synostosis, mutations in FGFR genes were found for all 38 patients with a syndromic form of craniosynostosis. Among 19 patients with nonsyndromic bilateral craniosynostosis, mutations were found in or near exon 9 of FGFR2 in 4 patients, as well as the common Pro250Arg mutation being found in exon 7 of FGFR3 in 10 patients; only 5 patients (9 percent) lacked a detectable mutation in FGFR 1, 2, or 3 [Mulliken et al., 1999]. This study suggests that mutation analysis should be considered in most patients with bilateral coronal synostosis.
Kleeblattschadel (Cloverleaf Skull)
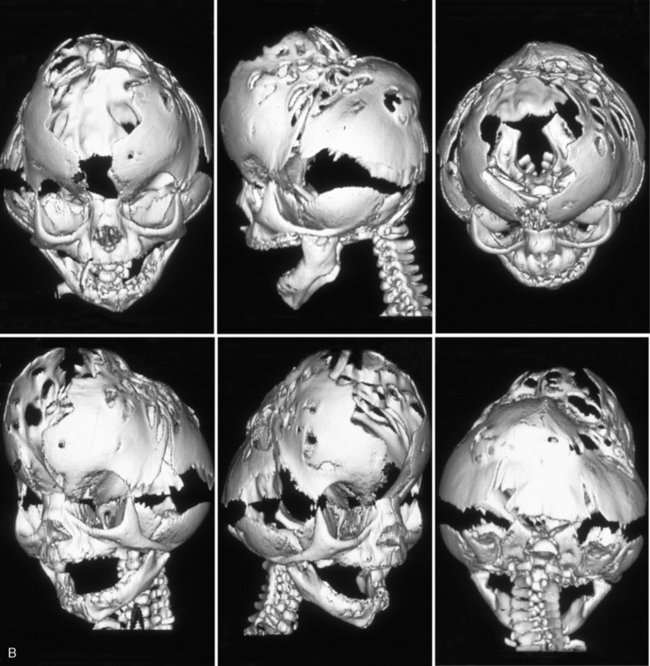
Kleeblattschadel is a term used to describe a cloverleaf skull configuration consisting of protrusion of each of the cranial bones, with broadening of the temporal region and face. These cranial protrusions are separated into focal bulges by furrows along the suture lines. The eyes often protrude, leading to corneal ulceration, scarring, and subsequent blindness, if the corneal surface remains unprotected [Stevenson, 1986]. Occipital encephaloceles can occur, and associated hydrocephalus is common. The palate is usually highly arched, but clefting is rare. The presence of Kleeblattschadel indicates that multiple sutural fusions occurred during early prenatal life. Increased thickening of the base of the occipital bone prevents lengthening of the skull, thus yielding the typical shape [Dambrain et al., 1987]. One child has also been described who had a cloverleaf skull without craniosynostosis (in which the primary defect was thought to be a cranial bone dysplasia that allowed for eventration of the brain and resultant cloverleaf configuration) [DR, 1988].
Multiple sutural synostosis is much more likely to result from genetic mutations in FGFR genes, TWIST, or MSX2, all of which result in syndromes which can present with cloverleaf skull [Cohen and MacLean, 2000]. The most common syndrome associated with cloverleaf skull is thanatophoric dysplasia type 2, which is due to mutations in FGFR3. Type 2 Pfeiffer’s syndrome, due to mutations in FGFR2, can result in cloverleaf skull, as can Crouzon’s syndrome and Apert’s syndrome, which are also due to mutations in FGFR2. Other syndromes, like FGFR3-associated coronal synostosis, rarely result in cloverleaf skull, but some rare syndromes like Boston craniosynostosis and Crouzon’s syndrome with acanthosis nigricans can sometimes manifest cloverleaf skull. In Boston craniosynostosis, the mutant MSX2 product has enhanced affinity for binding to its DNA target sequence, resulting in activated osteoblastic activity and aggressive cranial ossification [Warman et al., 1993; Jabs et al., 1993; Ma et al., 1996]. In Crouzon’s syndrome with acanthosis nigricans, a specific FGFR3 mutation (Ala391Glu), leads to early onset of acanthosis nigricans during childhood, often with associated choanal atresia and hydrocephalus [Schweitzer et al., 2001].
Eighty-five percent of children with Kleeblattschadel will have other anomalies, and the pattern is often consistent with a syndrome diagnosis. The prognosis is usually syndrome-dependent and can be quite poor, with early death due to respiratory difficulties or progressive brain damage from hydrocephalus being common [Frank et al., 1985]. However, subtotal craniectomy within the first 3 weeks of life in individuals with mild Kleeblattschadel may result in normal or near-normal development [Frank et al., 1985; Kroczek et al., 1986; Turner and Reynolds, 1980]. Early extensive calvariectomy is merited to preserve brain function and development, as well as to allow reformation of the craniofacial skeletal features. Many craniofacial surgeons prefer to begin with a posterior skull release in the early months of life (mean age 4 months), followed by fronto-orbital advancement around the end of the first year (mean age 14 months), with insertion of a ventriculoperitoneal shunt at the time of the first procedure if there is associated hydrocephalus [Sgouros et al., 1996]. The use of postoperative orthotic molding can help to channel brain growth into a more normal form, leading to improved postoperative results over those obtained via surgery alone [Schweitzer et al., 2001]. When lambdoid synostosis occurs as part of a syndrome with multiple suture involvement, there is often bilateral involvement, and early posterior release may alleviate some associated increased intracranial pressure. Patients with Kleeblattschadel need to be followed carefully for hydrocephalus, which may be part of the syndrome, rather than due to the multiple suture synostosis. Restricted growth of the posterior fossa is particularly common in severe craniofacial dysostosis syndromes. The purpose of surgery should be to decompress the brain, expand the bony orbits to accommodate the globes, and open airway passages [Kroczek et al., 1986].
Treatment and Outcomes of Craniosynostosis
Mild degrees of craniosynostosis may not always require surgery; however, in moderately severe cases, early surgery is usually warranted. The usual indication for surgery is to restore normal craniofacial shape and growth. When both the coronal and sagittal sutures are synostotic, impairing brain growth early in infancy, surgery is indicated to help prevent neurological and ophthalmologic complications associated with increased intracranial pressure and inadequate orbital volume. A variety of neurosurgical techniques have been developed for the treatment of craniosynostosis [Cohen and MacLean, 2000]. Most of these techniques involve removing the aberrant portion of the bony calvarium from its underlying dura, including the area surrounding the synostotic suture(s). If this is done within the first few months after birth, a new bony calvarium will usually develop within the remaining dura mater under the same principles that guide normal prenatal calvarial morphogenesis. As long as there is continued growth stretch from the expanding brain, the sites over the dural reflections remain unossified, thereby maintaining the sutures in a fibrous, open state. Thus, the calvarium and its sutures usually re-form normally after a partial calvariectomy for craniosynostosis. The new bony calvarium begins to develop within 2–3 weeks after surgery, and is usually firm by 5–8 weeks after surgery. If the procedure is done after 3–4 months of age, the approach is similar, with the exception that pieces of the calvarium are usually replaced in a mosaic pattern over the dura mater to act as niduses for the mineralization of new calvarium. Newer endoscopic repair techniques have been developed, followed by postoperative orthotic molding. Such procedures are most effective if done relatively early in infancy. These techniques are most effective in normal infants without a syndromic type of craniosynostosis.
Following early surgery for isolated craniosynostosis (primarily fronto-orbital advancement and/or calvarial vault remodeling at a mean age of 8 months), only 13 percent of 104 patients (10 bilateral coronal, 57 unilateral coronal, 29 metopic, and 8 sagittal) required a second cranial vault operation for residual defects at a mean age of 23 months. Perioperative complications were minimal (5 percent), with 87.5 percent of patients considered to have satisfactory craniofacial form, and low rates of hydrocephalus (3.8 percent), shunt placement (1 percent), and seizures (2.9 percent). Among such cases of isolated craniosynostosis, unilateral coronal synostosis was the most problematic type due to vertical orbital dystopia, nasal tip deviation, and altered craniofacial growth problems with residual craniofacial asymmetry [McCarthy et al., 1995]. In a second study of 167 children with both nonsyndromal (isolated) and syndromal craniosynostosis (12 bilateral coronal, 18 unilateral coronal, 39 metopic, and 46 sagittal), repeat operations were necessary in only 7 percent. Repeat operations were more common in syndromic cases (27.3 percent) than in nonsyndromic craniosynostosis (5.6 percent) [Williams et al., 1997].
Even though intracranial pressure can be elevated in patients with nonsyndromic craniosynostosis, they may not have decreased cranial volumes either before or after surgical repair, and as a group show slightly larger intracranial volumes when compared with normal controls. This could reflect the impact of fetal head constraint on fetuses with larger heads, or it might relate to the known association of macrocephaly with nonsyndromic coronal craniosynostosis due to the common Pro250Arg mutation in FGFR3 (which was not analyzed in these studies) [Polley et al., 1998]. Hydrocephalus occurs in 4–10 percent of patients with craniosynostosis and is more frequent with syndromic and multiple sutural craniosynostosis. In nonsyndromic patients, the rate of cerebral ventricular dilatation is the same as that observed in the general population, and it appears to be related to venous hypertension induced by jugular foramen stenosis. Such dilatation usually stabilizes spontaneously and rarely requires shunting. Some cases of progressive hydrocephalus in syndromic craniosynostosis cases were related to multiple sutural involvement, thereby constricting cranial volume, constricting the skull base, crowding the posterior fossa, and causing jugular foraminal stenosis [Cinalli et al., 1998]. These findings were most frequent among patients with Crouzon’s, Pfeiffer’s, and Apert’s syndrome, especially in association with cloverleaf skull abnormalities. A diffuse beaten copper pattern on skull radiographs, along with obliteration of anterior sulci or narrowing of basal cisterns in children under the age of 18 months, is predictive of increased intracranial pressure in over 95 percent of cases [Tuite and Lindquist, 1996].
Nonsyndromic Craniosynostosis Neurocognitive Development
There is a growing body of evidence that single-suture craniosynostosis is associated with neurobehavioral problems [Kapp-Simon, 1998; Magge et al., 2002]. In the 2004 manuscript, Speltz et al. nicely summarized all of the published neurobehavioral studies conducted on children with single suture craniosynostosis [Speltz et al., 2004].
Few studies have documented actual rates of mental retardation (typically defined as standardized test scores below 70). Several older studies reported slightly increased rates of mental retardation (from 6.5 to 12 percent) in comparison to an expected rate of about 2.2 percent in the population [Kapp-Simon, 1998; Hunter and Rudd, 1976, 1977; Sidoti et al., 1996].
Syndromes with multiple-suture fusions have more commonly been associated with elevated rates of mental retardation and learning disabilities [Cohen, 1991]. Most studies have found adverse neurocognitive outcomes in about 35–40 percent of assessed cases [Bottero et al., 1998; Shimoji et al., 2002; Shipster et al., 2003], with an occasional study finding this number to be as high as 50 percent [Kapp-Simon, 1998; Magge et al., 2002]. A few studies have reported a 3–5 times higher than average risk of poor neurobehavioral outcome. In studies that directly measured the IQs of children with sagittal synostosis, a discrepancy of greater than 20 standard score points between language and nonverbal IQ scores was found [Magge et al., 2002; Shipster et al., 2003]. In 2002, Magge et al. reported a 50 percent incidence of learning disability and found that verbal IQ was significantly higher than nonverbal IQ.
Sagittal synostosis has not been found to affect the neurological function of the infant or toddler significantly [Kapp-Simon et al., 1993]. However, older children may demonstrate moderately severe speech and language difficulties in up to 37 percent of cases, and the tendency toward such problems is associated with a positive family history for such difficulties and later age of surgical correction [Shipster et al., 2003; Virtanen et al., 1999; Panchal et al., 1999]. In a study of 30 older untreated children (average age 9.25 years) with sagittal synostosis, almost all patients and parents were pleased with their decision and patients demonstrated normal cognitive and school performance, as well as normal behavior and psychological adjustment on standardized testing [Boltshauser et al., 2003].
In a long-term neurodevelopmental outcome study of nonsyndromic cases, a correlation was found between outcomes and timing of surgery, with 22.6 percent of those operated on before 12 months showing impaired mental development, versus 52.2 percent impaired mental development in those operated on after age 12 months. Overall, 31 percent of trigonocephaly cases had delayed development, and this appeared to be related to the severity of the problem and the presence of associated malformations [Bottero et al., 1998].

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


