Chapter 94 Metabolic Myopathies
Utilization of Bioenergetic Substrates in Exercise
Defects of energy metabolism may profoundly disrupt the function of muscle and other highly energy-dependent tissues, such as brain, nerve, heart, kidney, liver and bowel. The limits of energy utilization in skeletal muscle are set by the adenosine triphosphatases (ATPases) that couple muscle contraction (myosin ATPase) and ion transport (calcium and sodium, potassium ATPases) to the hydrolysis of ATP to adenosine diphosphate (ADP) and inorganic phosphate (Pi) [Kushmerick, 1995]. ADP and Pi in turn activate energy-producing reactions that regenerate ATP. Without this, ATP stores would be exhausted in seconds. The substrates that are used to replenish ATP are determined by the intrinsic properties of these fuels and by the intensity and duration of exercise that modulates fuel selection [Gollnick, 1985; Astrand and Rodahl, 1986]. The creatine kinase reaction and anaerobic glycogenolysis are the major anaerobic sources of ADP phosphorylation. Increases in ADP and adenosine monophosphate (AMP) that occur in heavy exercise are primarily buffered by the coupled adenylate kinase (myokinase), adenylate deaminase (myoadenylate deaminase) reactions. Anaerobic glycogenolysis and phosphocreatine hydrolysis support muscle energy production with 2- to 4-fold higher rates than those supported by oxidative metabolism [Sahlin, 1986]. Anaerobic energy is crucial for rapid bursts of exercise and to fuel the transition from rest to exercise. The acceleration to high rates of energy production occurs instantly for ATP, in less than a second for phosphocreatine, and within seconds for anaerobic glycogenolysis. In contrast, maximal oxidative power requires from 3 minutes (with glycogen as the oxidative substrate) to 30 minutes (for peak fatty acid oxidation). Anaerobic fuels are rapidly depleted and lead to the accumulation of metabolic end products, such as protons and Pi, that promote fatigue.
If exercise needs to be sustained for more than a few minutes, then oxidative phosphorylation is necessary and provides the most abundant source of ATP synthesis. Glycogen is the major endogenous oxidative fuel of skeletal muscle, while blood glucose and free fatty acids (FFA) are the major exogenous fuels. A small percentage of muscle energy needs is supplied by amino acids, predominantly branched chain amino acids, which are oxidized to a limited extent. Oxidative metabolism provides higher yields of ATP per mole of substrate, rising from 2 to 36 for glucose and from 3 to 37 per glycosyl unit of glycogen metabolized anaerobically versus oxidatively. Furthermore, the metabolic end products of oxidative metabolism – namely, CO2 and water – are removed easily from working muscle and do not promote fatigue. The most abundant and critical fuel for the support of prolonged, moderate exercise is lipid. Carbohydrate stores, in the form of muscle and hepatic glycogen and blood glucose (derived mainly from hepatic glycogenolysis), are limited and can support high-intensity exercise for only 1–2 hours. However, carbohydrate, particularly muscle glycogen, is critical for normal oxidative metabolism. Glycogen supports a peak rate of oxidative phosphorylation that is about 2-fold greater than fat. Though incompletely understood, this may be based upon a requirement for glycogen-derived pyruvate to support optimal function of the tricarboxylic acid cycle [Sahlin et al., 1990, 1995; Gibala et al., 1997]. The proportion of carbohydrate relative to lipid oxidation increases progressively as the intensity of the aerobic exercise increases, until carbohydrate is the exclusive fuel of maximum oxidative metabolism [Sahlin, 1986; van Loon et al., 2001]. Second, glycogen is able to accelerate to maximal oxidative power output rapidly compared to other fuels [Sahlin, 1986; Haller and Vissing, 2002]. Third, the molar ratio of ATP produced to O2 consumed is higher for glycogen (6.17) and glucose (5.98) than for fatty acids (5.61) [Rennie and Edwards, 1981]. The importance of this point lies in the fact that peak O2 utilization in healthy humans is limited by O2 delivery [Saltin, 1988].
The primary source of energy for resting muscle is derived from fatty acid oxidation (FAO) [Felig and Wahren, 1975]. At rest, glucose utilization accounts for 10–15 percent of total oxygen consumption [Wahren, 1977]. Both slow- and fast-twitch fibers have similar levels of glycogen content at rest [Essen, 1978]. The choice of the bioenergetic pathway in working muscle depends not only on the type, intensity, and duration of exercise [Essen, 1977; Gollnick et al., 1974], but also on diet and physical conditioning [DiMauro and Haller, 1999]. In the first 5–10 minutes of moderate exercise, high-energy phosphates are used first to regenerate ATP. This is followed by muscle glycogen breakdown, which is indicated by a sharp rise in lactate during the first 10 minutes. Blood lactate levels then drop as muscle triglycerides and blood-borne fuels are used [Felig and Wahren, 1975; Lithell et al., 1979]. After 90 minutes, the major fuels are glucose and FFAs. During 1–4 hours of mild to moderate prolonged exercise, muscle uptake of FFAs increases approximately 70 percent, and after 4 hours, FFAs are used twice as much as carbohydrates.
Box 94-1 Heritable Causes of Myoglobinuria
I Biochemical Abnormality Known
III Abnormal Composition of Sarcolemma
(Modified from Tein I, et al: In Rowland LP, DiMauro S, editors: Handbook of clinical neurology, vol 18[62], Myopathies, Amsterdam, 1992, Elsevier Science Publishers.)
Myoglobinuria
Myoglobinuria is a clinical syndrome, not just a biochemical state [Rowland, 1984]. In the alert patient, myalgia and limb weakness are the most common presenting symptoms. Urine color is usually brownish rather than red, and the urine tests positive for both albumin and heme (a concentration of at least 4 μg/mL). There are few or no red blood cells. Myoglobin can be identified by immunochemical methods. The sarcoplasmic enzymes, including serum creatine kinase (CK), are usually elevated to more than 100 times normal. Inconstant features include hyperphosphatemia, hyperuricemia, and hypocalcemia. If renal failure occurs, serum potassium and calcium levels may rise. If the patient is comatose or if the presenting disorder is one of acute renal failure, there may be no muscle symptoms or signs. Under these conditions, the diagnosis can be made if:
The etiologies of heritable myoglobinuria differ in adults compared with children. In a study of 77 adult patients aged 15–65 years, Tonin et al. [1990] identified the enzyme abnormality in 36 patients (47 percent) as follows: CPT deficiency in 17 patients; glycolytic defects in 15 patients (including PPL in 10, PPL b kinase in 4, and PGK in 1); myoadenylate deaminase in 3; and combined CPT and myoadenylate deaminase in 1. In contrast, in 100 cases of recurrent childhood-onset myoglobinuria, a lower percentage of children have been diagnosed biochemically (24 percent): 16 with CPT deficiency, 1 with SCHAD deficiency, and 7 with various glycolytic defects, including 2 PPL, 1 PGK, 3 PGAM, and 1 LDH deficiency [Tein et al., 1990b]. These children could be divided into two groups – a type I exertional group, in which exertion was the primary precipitating factor (56 cases); and a type II toxic group, in which infection and/or fever and leukocytosis were the primary precipitants (37 cases). The type II toxic childhood group was distinguished from the type I exertional childhood- and adult-onset groups by its etiologies, which were limited to FAO defects, as well as its slight female predominance, which contrasted with the marked male predominance in the latter two groups. The type II toxic group was distinguished further by the earlier age at onset of myoglobinuria, the presence of a more generalized disease (e.g., ictal bulbar signs, seizures, encephalopathy, developmental delay), and a higher mortality rate. Currently, the most common etiology for recurrent myoglobinuria in both adults and in children is CPT II deficiency [Tein et al., 1992].
Glycogenoses
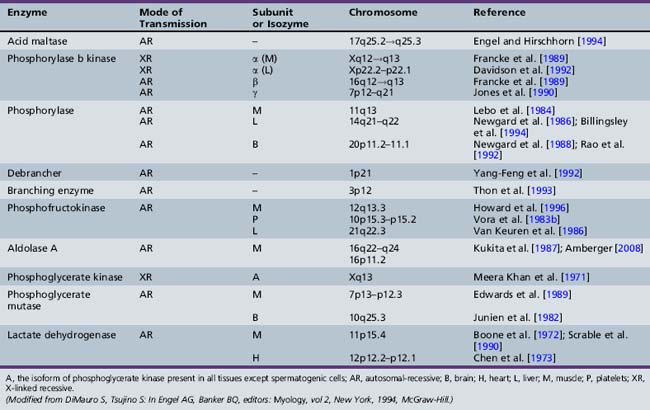
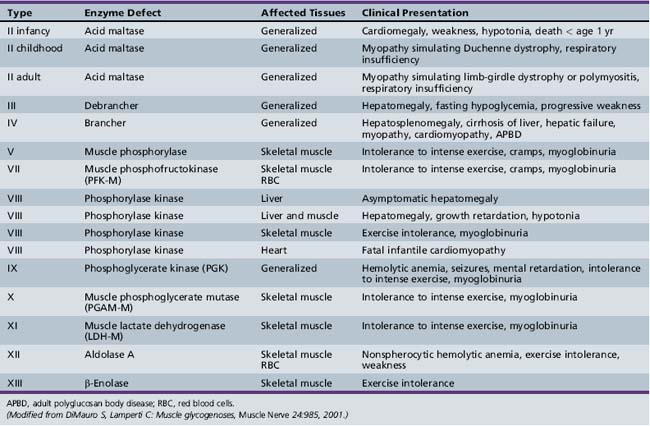
Only glycogenoses affecting skeletal muscle, alone or in association with other tissues, are discussed in this section (Figure 94-1) [DiMauro and Bresolin, 1986]. Molecular genetic analysis has led to the cloning of genes encoding the enzymes involved in most glycogenoses, as well as their chromosomal localization (Table 94-1). All are autosomal-recessive in inheritance, with the exception of PGK, which is X-linked recessive, and PPL b kinase, which may be either. In the glycogenoses, fixed weakness is the primary presentation in acid maltase, debrancher, aldolase, and brancher enzyme deficiencies, whereas exercise intolerance with cramps, myalgia, and recurrent myoglobinuria is the primary presentation in phosphorylase kinase, phosphorylase, phosphofructokinase, phosphoglycerate kinase, phosphoglycerate mutase, β-enolase, and lactate dehydrogenase deficiencies (Table 94-2) [DiMauro and Lamperti, 2001].
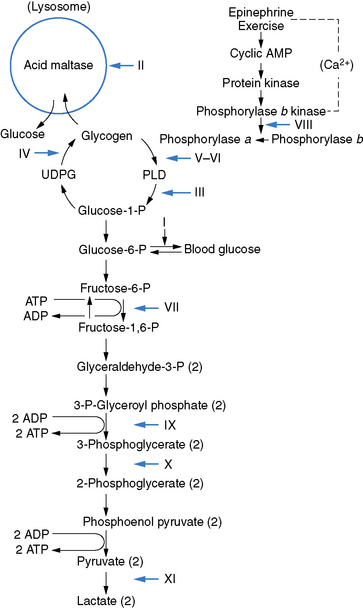
Fig. 94-1 Scheme of glycogen metabolism and glycolysis.
(From DiMauro S, et al: Metabolic myopathies, Am J Med Genet 25:635–651, 1986.)
Pathophysiology
The immediate source of energy for contraction and relaxation is provided by the hydrolysis of ATP. Oxidative phosphorylation provides the largest contribution of energy overall, whereas anaerobic glycolysis plays a relatively minor role, primarily limited to conditions of sustained isometric contraction when blood flow and oxygen delivery to exercising muscles are reduced drastically. The dynamic form of exercise, such as walking or running, is primarily dependent on aerobic glycolysis. Therefore, the pathophysiology of glycogenoses relates more to the impairment of aerobic than anaerobic glycolysis [Lewis and Haller, 1986; Lewis et al., 1991].
The energy substrates used by muscle for aerobic metabolism depend on the type, intensity, and duration of exercise, as well as on physical conditioning and diet. During intense exercise (close to maximal oxygen uptake, or VO2max, in dynamic exercise or maximal force generation in isometric exercise), energy is derived from anaerobic glycolysis, particularly when there is a burst of activity with rapid acceleration to maximal exercise [DiMauro and Tsujino, 1994]. During low-intensity exercise (below 50 percent VO2max), the primary source of energy is derived from blood glucose and FFAs. At higher intensities, the proportion of energy derived from carbohydrate oxidation increases, and glycogen becomes an important fuel. At 70–80 percent of VO2max, the critical energy source is provided by the aerobic metabolism of glycogen, and fatigue occurs when glycogen stores are exhausted [DiMauro and Tsujino, 1994]. Individuals with defective glycolysis/glycogenolysis are most vulnerable during the initial stages of intense exercise, and they must rest soon after beginning exercise because of muscle cramps. However, if they continue to exercise at low intensity, they are able to continue for a longer time. This is known as the second-wind phenomenon, and has been attributed to a metabolic switch from carbohydrate to fatty acid utilization [Felig and Wahren, 1975], and to increased circulation with increased availability of blood glucose from hepatic glycogenolysis (Haller et al., 1985]. A decrease in ATP levels could first cause muscle contracture. Theoretically, a more severe depletion could lead to myoglobinuria [Rowland, 1984], although this has not been proven.
The forearm ischemic exercise test, developed by McArdle [1951], is a useful test for the detection of enzymatic defects in the nonlysosomal glycogenolytic and glycolytic pathways. This test can be performed in cooperative children as young as 6 years of age. A catheter is placed in a superficial antecubital vein, and basal lactate and ammonia levels are obtained without stasis. A sphygmomanometer cuff is then placed above the elbow and inflated above arterial pressure. The patient is asked to squeeze another rolled-up cuff rhythmically to well above 120 mmHg for 1 minute of exercise. This requires constant encouragement from the observer because significant discomfort can occur, even in healthy control subjects. The test should be truncated if the patient develops an acute cramp, as myonecrosis and/or a compartment syndrome may occur in an individual with a glycolytic disorder [Meinck et al., 1982; Lindner et al., 2001]. After 1 minute of exercise, the cuff around the arm is deflated and blood samples are sequentially obtained at 1, 3, 5, 7, 10, and 15 minutes. In healthy subjects there is a 4- to 6-fold increase of lactate over baseline, with the peak occurring at 1–2 minutes post exercise, which declines to baseline values by 15 minutes. This is paralleled by a similar 5-fold or more increase in ammonia, with levels generally peaking at 2–5 minutes after exercise in individuals with normal myoadenylate deaminase levels. In healthy subjects, venous ammonia and lactate are linearly related [Sinkeler et al., 1985]. In individuals with a defect in glycolysis/glycogenolysis, there is an insufficient rise in lactate (less than 2-fold), with a compensatory and exaggerated increase in ammonia, which also indicates sufficient effort on the part of the individual. This exaggerated rise in ammonia is attributable to high cellular levels of ADP, resulting from a combination of blocked glycogenolysis/glycolysis and absent cellular acidosis. An insufficient lactate rise has been demonstrated in PPL, debrancher, PFK, PGK, PGAM, and LDH deficiencies but not in acid maltase or phosphorylase b kinase deficiency. The major limitation of this test is that the rise of venous lactate in individuals who do not have a defect in this pathway is highly dependent on the patient’s ability and willingness to exercise. Therefore, patients in whom lactate levels are low, due to poor effort or to placement of the venous line in other than the median cubital vein, show proportionally blunted ammonia responses.
Given the potential risk of myoglobinuria, alternatives to the traditional ischemic forearm test have been described. One of these employs sustained, intense (70 percent of maximal voluntary contraction, MVC) isometric handgrip exercise [Hogrel et al., 2001]. However, this form of testing is as ischemic as the use of a blood pressure cuff, since the intramuscular pressure in isometric contractions of ≥50 percent MVC completely occludes muscle blood flow. Therefore, any reduction in the incidence of muscle contractures is dependent upon shortening the duration of the test that is also effective in minimizing contractures in the traditional ischemic forearm test. An alternative nonischemic forearm test has been described and involves 30 maximal handgrip contractions in 1 minute without a blood pressure cuff [Kazemi-Esfarjani et al., 2002]. The level of increase in lactate and ammonia in control subjects and the diagnostic sensitivity in patients were similar to the ischemic exercise test. However, in contrast to the ischemic tests, the retained oxidative capacity helps to protect individuals from contractures or significant pain [Kazemi-Esfarjani et al., 2002].
Glycolytic/Glycogenolytic Defects
Acid Maltase Deficiency
Clinical features
Acid maltase deficiency (AMD, glycogenosis type II, GSD type II) results in three very different clinical presentations [DiMauro and Lamperti 2001]:
Slonim et al. [2000] have described a subgroup of patients who share muscle morphological features with the neonatal-onset infantile form patients, but differ from typical Pompe’s disease patients in that the heart is not involved and survival is longer.
Laboratory data
All forms of AMD demonstrate an increase in serum CK. Electromyography (EMG) reveals myopathic abnormalities and may also show fibrillation potentials, positive waves, complex repetitive discharges, and myotonic discharges [DiMauro and Lamperti, 2001]. These are more frequently demonstrated in the paraspinal muscles. The electrocardiogram (EKG) demonstrates characteristic, although not specific, changes, including a short PR interval, giant QRS complexes, and signs of biventricular hypertrophy in infantile AMD. In adults, there is markedly reduced vital capacity.
Pathology
In infantile AMD, glycogen accumulation is evident in all tissues, most notably the heart. There is significant involvement of the anterior horn cells of the spinal cord and of brainstem nuclei, leading to severe weakness and fasciculations. Glycogen accumulation in Schwann cells is seen in peripheral nerve biopsies. There is a vacuolar myopathy on muscle biopsy in all three forms of AMD [DiMauro et al., 1992]. In the infantile form, all muscle fibers contain many vacuoles, which often coalesce into a lacework pattern and contain periodic acid–Schiff (PAS)-positive, diastase-digestible material and stain for acid phosphatase. Electron microscopy reveals excess glycogen either within lysosomal vacuoles or free in the cytoplasm.
Biochemistry and molecular genetics
Although the enzyme defect is generalized and can be documented in lymphocytes [Shanske and DiMauro, 1981] or in cultured skin fibroblasts, biochemical measurement of acid maltase in muscle establishes the diagnosis. It is not entirely clear why symptoms are mainly confined to muscle in the childhood and adult forms. However, differences in clinical expression may relate, in part, to the amounts of residual activity [Mehler and DiMauro, 1977; Reuser et al., 1987; Van Der Ploeg et al., 1988]. Second, genetic heterogeneity has also been demonstrated. In a study of 14 patients, Martiniuk et al. [1990a, 1990b] found that messenger RNA (mRNA) was lacking in 5 of 10 infantile AMD cases and was present in all 4 adult cases, although it was shorter in 2 cases. The defective lysosomal enzyme, α-glucosidase, is encoded by a gene on chromosome 17 [D’Ancona et al. 1979; Weil et al., 1979; Solomon et al., 1979], and over 55 mutations have been identified in patients with the three variants [DiMauro and Lamperti, 2001]. The genotype–phenotype correlation is hard to establish due to the frequent compound heterozygosity. However, there is good correlation between the severity of the mutation and the severity of the clinical phenotype. Therefore, deletions and nonsense mutations are usually associated with the infantile variant, whereas “leaky” mutations, such as the IVS1(-13T>G) splice-site mutations, are associated with the adult-onset variant [Hirschhorn, 1995]. Childhood AMD is often the result of compound heterozygosity [Huie et al., 1998], but has also been associated with at least one homozygous, apparently specific mutation [Adams et al., 1997]. A reliable and rapid first-tier test for screening patients suspected of having Pompe’s disease has been provided, using a fluorometric dried blood spot-based α-glucosidase activity assay [Goldstein et al., 2009]. The measurement of acid α-glucosidase activities in dried blood spots using tandem mass spectrometry has been found suitable for high-throughput analysis and newborn screening for Pompe’s disease [Dajnoki et al., 2008].
Treatment
Clinical trials with lysosome-stabilizing agents and activators of glycogenolysis have been unsuccessful in Pompe’s disease [DiMauro et al., 1992]. Recent experimental work with small molecule pharmacological chaperones may hold promise for future clinical trials [Flanagan, 2009]. Enzyme replacement looks promising, particularly for patients with childhood and adult AMD. Recombinant human α-glucosidase, obtained from milk of transgenic rabbits and injected into knockout mice lacking acid maltase, corrected the enzyme defect fully in all tissues but brain [Bijvoet et al., 1999]. Amalfitano et al. [2001] reported the results of a phase I/II open-label, single-dose study of recombinant human α-glucosidase infused intravenously twice weekly in three infants with infantile GSD type II. The results of more than 250 infusions showed that recombinant human α-glucosidase was generally well tolerated. There were steady decreases in heart size and maintenance of normal cardiac function for more than 1 year observed in all three infants. These infants lived past the critical age of 1 year and also show improvements in skeletal muscle function. In a 52-week trial with acid glucosidase alpha, there was marked improvement in the cardiomyopathy, ventilatory function, and overall survival among 18 children under 7 months of age with infantile Pompe’s disease [Kishnani et al., 2009]. Sixteen of the 18 patients enrolled in an extension study continued to receive alglucosidase alpha for up to a total of 3 years and showed reduction in the risk of death by 95 percent, and reduced risk of invasive ventilation or death by 91 percent, compared to an untreated historical control group, over the entire study period. The cardiomyopathy continued to improve and 11 patients learned and sustained substantial motor skills. In another study of late-onset AMD, three patients (aged 11, 16, and 32 years), all wheelchair-dependent and two ventilator-dependent with a history of deteriorating pulmonary function, received 3 years of treatment with weekly infusions of recombinant α-glucosidase from rabbit milk, and had stabilized pulmonary function and reported less fatigue [Winkel et al., 2004]. The youngest and least affected patient showed significant improvement of skeletal muscle strength and function, and, after 72 weeks of treatment, could walk without support. There have also been promising gene therapy results, both in vitro and in vivo, using E1-deleted recombinant adenovirus encoding human α-glucosidase. Transduction of the viral construct into enzyme-deficient human fibroblasts in culture resulted in a dose-dependent return of acid maltase activity, which was localized within lysosomes [Pauly et al., 1998]. Equally encouraging results were obtained by injecting a similar viral construct into the pectoral muscle of acid maltase-deficient Japanese quails that are an avian model of AMD [Tsujino et al., 1998]. Individuals with childhood or adult AMD may require ventilatory assistance. A high-protein diet has improved strength and respiratory function in some but not all patients [DiMauro et al., 1992; Slonim et al., 1983].
Phosphorylase b Kinase Deficiency
Clinical features
Phosphorylase b kinase (PBK) deficiency (glycogenosis type VIII) may be divided into four clinical syndromes [DiMauro and Tsujino, 1994]. These can be differentiated by inheritance and tissue distribution as follows:
Laboratory data
The resting serum CK is variably increased in most patients. The EMG may be normal or may show nonspecific myopathic changes. Muscle biopsy may be normal or show subsarcolemmal accumulations of glycogen, primarily located in type 2B fibers. The PPL stain is normal. There are pools of free, normal-looking glycogen particles on electron microscopy [DiMauro and Tsujino, 1994].
Biochemistry and molecular genetics
PBK participates in glycogen metabolism regulation by acting on the two main enzymes involved in glycogen synthesis and degradation: namely, glycogen synthetase and PPL [Heilmeyer, 1991]. PBK converts PPL from the less active b form to the more active a form, and simultaneously converts glycogen synthetase from a more active, dephosphorylated form to a less active, phosphorylated form. Thus, when glycogen synthesis is turned on, glycogen degradation is turned off, and vice versa. PBK is a multimeric enzyme composed of four different subunits (alpha, beta, gamma, delta) [DiMauro and Tsujino, 1994]. The alpha and beta subunits are regulatory, the gamma subunit is catalytic, and the delta subunit is identical to calmodulin and confers calcium sensitivity to the enzyme [DiMauro and Lamperti, 2001]. In addition, there are two isoforms for the alpha subunit (muscle and liver), which are encoded by genes on the X chromosome. The two isoforms for the gamma subunit (muscle and testis) and the beta subunit are encoded by autosomal genes. Glycogen is normal or moderately increased in patients with myopathy, and the PBK activity is absent or markedly decreased [DiMauro and Tsujino, 1994].
It is not easy to account for the tissue specificity in the different clinical variants. Two distinct genes have been identified. One gene (PHKA1) is on the proximal long arm at Xq13, which encodes a muscle isozyme; the other gene (PHKA2) is on the distal short arm of the X chromosome at Xp22.2–p22.1, which encodes a nonmuscle isozyme, and is in the same region to which the gene for X-linked liver glycogenosis resulting from PBK deficiency has been mapped [Willems et al., 1991]. Tissue-specific isozymes may be the result of alternative RNA splicing. DiMauro and Tsujino [1994] suggest that mutations in tissue-specifically expressed exons of the beta subunit [Harmann et al., 1991] could explain the apparently autosomal-recessive forms of myopathy and fatal infantile cardiomyopathy.
Phosphorylase Deficiency
Clinical features
Myophosphorylase deficiency (type V glycogenosis, or McArdle’s disease) is a rare disease but is an important cause of recurrent myoglobinuria. Exercise intolerance generally starts in childhood, but overt episodes of muscle cramping and myoglobinuria develop later in adolescence. Brief isometric contraction (e.g., lifting heavy objects) and less intense but sustained dynamic exercise (e.g., climbing stairs) are the two primary precipitants. Approximately 50 percent of these patients experience episodes of muscle necrosis and myoglobinuria after exercise, while 27 percent develop acute renal failure. Although there is usually complete functional recovery after the episode of myoglobinuria, about one-third of patients have fixed mild proximal greater than distal weakness, which is seen more commonly in older patients [DiMauro and Bresolin, 1986]. The diagnosis is usually made in the second or third decade of life. There may be a marked variation in the severity of symptoms. In some, progressive weakness may begin late in life (sixth decade), with no history of cramps or pigmenturia [DiMauro and Bresolin, 1986; Pourmand et al., 1983]. The cumulative effect of recurrent muscle damage over the years may explain the appearance of fixed weakness in older individuals. In contrast, there have been four unique childhood cases with severe generalized muscle and respiratory weakness noted at or soon after birth, with death in infancy [DiMauro and Hartlage, 1978; DiMauro and Tsujino, 1994; Milstein et al., 1989]. A hypotonic 1-year-old infant has been described with repeated episodes of hyper-creatine kinase-emia during febrile episodes [Ito et al., 2003].
Laboratory data
Between episodes of myoglobinuria, the serum CK is variably increased in 93 percent of cases. This serves as a differentiating feature from CPT deficiency, in which the resting CK generally is normal. EMG between episodes of myoglobinuria demonstrates fibrillations, myotonic discharges, and positive waves in up to 50 percent of patients, suggesting a mild myopathy [DiMauro and Tsujino, 1994]. There is a lack of cytoplasmic acidification on phosphorus-31 nuclear magnetic resonance (31P-NMR) spectroscopy during aerobic or ischemic exercise, as well as a greater than normal drop of the phosphocreatine/inorganic phosphate ratio [Argov and Bank, 1991; Ross et al., 1981].
Inheritance
There is an unexplained marked male predominance, despite the autosomal-recessive pattern of inheritance. The muscle isoform gene has been localized to chromosome 11 [Lebo et al., 1984]. Two families with apparent autosomal-dominant transmission [Chui and Munsat, 1976; Schimrigk et al., 1967] may be explained on the basis of subsequent generations of homozygotes and manifesting heterozygotes, in whom the residual activity was below a critical threshold [Papadimitriou et al., 1990; Schmidt et al., 1987]. In a third family, Tsujino et al. [1993a] demonstrated that the affected mother was a compound heterozygote carrying two different point mutations in the PPL gene, the unaffected father was heterozygous for a third point mutation, and the three affected children were compound heterozygotes.
Muscle biopsy
There may or may not be focal accumulations of glycogen between myofibrils and in the subsarcolemmal regions on PAS stain. The PPL stain [Takeuchi and Kuriaki, 1955] shows no staining in muscle fibers, in contrast to normal staining of the smooth muscle in the walls of intramuscular vessels. DiMauro and Tsujino [1994] pointed out that a positive histochemical reaction may be seen in McArdle’s disease under the following two conditions:
A false-positive reaction in regenerating fibers is due to expression of a different isozyme in immature muscle cells [DiMauro et al., 1978; Sato et al., 1977]. Accumulation of normal-looking glycogen beta particles under the sarcolemma and between myofibrils and myofilaments may be seen on electron microscopy [DiMauro and Tsujino, 1994].
Biochemical considerations
PPL (alpha-1,4-glucan orthophosphate glycosyl transferase) initiates glycogen breakdown by removing 1,4-glucosyl residues phosphorolytically from the outer branches of the glycogen molecule, with liberation of glucose-1-phosphate until the peripheral chains have been shortened to approximately four glucosyl units. The debranching enzyme catalyzes further breakdown. The combined action of these two enzymes degrades glycogen to glucose-1-phosphate (approximately 93 percent) and glucose (approximately 7 percent). Muscle PPL exists as an active phosphorylated alpha form and a less active dephosphorylated beta form. The muscle PPL activity is undetectable in most or up to 10 percent of normal residual activity in affected patients [DiMauro and Tsujino, 1994]. Glycogen accumulation is moderate (about 2-fold) or normal. The glycogen structure is normal. There is no enzyme protein in muscle immunologically detectable by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE), immunoblot, and enzyme-linked immunosorbent assay (ELISA) in the majority of patients [McConchie et al., 1991; Servidei et al., 1988b]. Normal mature human muscle has a single PPL isozyme, whereas cardiac muscle and brain have three different isozymes on PAGE, including:
Lack of the muscle isozyme therefore should cause partial defects in the heart and brain. A defect of the brain isozyme would be expected to cause heart and brain involvement [DiMauro and Tsujino, 1994].
Pathophysiology
The pathophysiology of exercise intolerance in McArdle’s disease involves a combination of interconnected factors related to impaired ATP production from both aerobic and anaerobic glycolysis. The decrease of ATP generated during anaerobic glycolysis may affect muscle ATPases selectively [James et al., 1996], including the Na+K+-ATPase. This inhibition, together with a decrease in Na+K+ pump numbers, may lead to excessive increases in blood and extracellular potassium, which typically are seen during exercise in McArdle’s disease [Haller et al., 1998a]. These changes can explain, in turn, the sarcolemmal inexcitability during repetitive neural stimulation [Dyken et al., 1967]. Furthermore, the lack of lactic acid production and intracellular acidification during exercise alters the equilibrium of the CK reaction, resulting in an exaggerated rise of ADP during exercise. The excessive intracellular ADP is converted to AMP, inosine monophosphate, ammonia, and adenine nucleotide degradation products, including inosine, hypoxanthine, and uric acid [Mineo et al., 1987]. Premature fatigue may be due to impaired excitation–contraction coupling, mediated by excessive accumulation of ADP [Lewis and Haller, 1991a]. Oxidative metabolism is also affected in McArdle’s disease because the block in glycogenolysis impairs production of pyruvate, which is a major anaplerotic substrate for the Krebs cycle [Sahlin et al., 1995]. In patients with McArdle’s disease, oxygen extraction and maximal oxygen uptake are decreased; these can be restored partially by administration of intravenous glucose [Haller et al., 1985].
Molecular genetics
The three PPL isozyme genes have been cloned, sequenced, and localized to different chromosomes [Billingsley et al., 1994; Lebo et al., 1984; Newgard et al., 1986, 1988; Rao et al., 1992] (see Table 94-1). Tsujino et al. [1993a] identified three different point mutations in the muscle isozyme gene: a C to G mutation in codon 49 of exon 1 (converting an arginine to a stop codon), a G to A mutation in codon 204 of exon 5 (converting a glycine to a serine), and an A to C mutation in codon 542 of exon 14 (converting a lysine to a threonine). In an analysis of 32 patients, 15 were homozygous for the first mutation and 12 were compound heterozygotes [Tsujino et al., 1993a]. More than 100 mutations have been identified in the muscle isozyme gene, which will facilitate carrier identification [DiMauro and Tsujino, 1994; Tsujino et al., 1993a, 1994a, 1994b, 1994c; DiMauro and Lamperti, 2001; Vissing et al., 2009]. These include missense, nonsense, and splice-junction mutations. The most common mutation in Europe and North America is the Arg49Stop, which accounts for 81 percent of the alleles in British patients [Bartram et al., 1993] and 63 percent of alleles in a survey of U.S. patients [El-Schahawi et al., 1996]. The high frequency of this mutation, on at least one allele, has facilitated molecular diagnosis of blood samples from patients with suspected McArdle’s disease. However, it is important to realize that the frequency of different mutations varies in different ethnic groups. The frequency of the Arg49Stop mutation appears to decrease from north to south in Europe, from 81 percent of alleles in the United Kingdom [Bartram et al., 1993] to 56 percent in Germany [Vorgerd et al., 1998] and 32 percent in Italy [Martinuzzi et al., 1996].
To date, there has been no clear genotype–phenotype correlation in McArdle’s disease. For example, the common homozygous Arg49Stop mutation was also present in an infant with the fatal myopathic variant [Tsujino et al., 1993a], and in a child who died of sudden infant death syndrome [El-Schahawi et al., 1997]. This lack of clear genotype–phenotype correlation was also documented in a study of 54 Spanish patients with McArdle’s disease from 40 unrelated families [Martin et al., 2001]. One possible explanation for increased severity of clinical presentation would be the presence of additional gene defects, such as myoadenylate deaminase deficiency [Rubio et al., 1997a; Tsujino et al., 1995a]. Recently, two patients with atypical McArdle’s disease, who carried common mutations on one allele and novel splice mutations in introns 3 and 5 on the other allele, demonstrated minimal phosphorylase activity that ameliorated the typical phenotype by augmenting muscle oxidative capacity [Vissing et al., 2009].
Treatment
A key therapeutic strategy has been the attempt to bypass the metabolic block by providing the muscle with glycolytic substrates [DiMauro and Tsujino, 1994]. The oral administration of glucose or fructose to raise the blood glucose level has been helpful but has resulted in weight gain. Glucagon injections have been impractical, with inconsistent results. Although exercise tolerance was increased by raising the serum FFA concentration through the use of fat emulsions, fasting, and the administration of norepinephrine or heparin, more practical regimens, such as a high-fat, low-carbohydrate diet, have not been effective [DiMauro and Bresolin, 1986]. In a study of intralipid infusion to increase FFA availability during 70 percent maximal oxygen consumption cycle ergometry, FFA levels more than tripled, but heart rate was significantly higher during exercise compared with placebo and glucose infusions trials, suggesting that, although lipids are an important source of fuel for exercising muscle in McArdle’s disease, the maximal rates of fat oxidation appeared limited and could not be increased above physiological normal rates during exercise [Andersen et al., 2009]. It was suggested that the limitation was probably caused by a metabolic bottleneck in the tricarboxylic acid cycle due to impaired glycolytic flux, and that therapies aimed at enhancing fat use in McArdle’s disease should be combined with interventions targeting expansion of the tricarboxylic acid cycle. An alternative strategy was to supply branched-chain amino acids, which are taken up, rather than released, by tissue of patients with McArdle’s disease during exercise [Wahren et al., 1973]. Slonim and Goans [1985] demonstrated an improvement of muscle endurance and strength in a patient with weakness after institution of a high-protein diet. Direct administration of branched-chain amino acids to six patients, however, resulted in impairment rather than improvement of bicycle exercise capacity in 5 of 6 patients, possibly due to a lowering of FFA by the amino acids [MacLean et al., 1998]. In a more recent study, patients with McArdle’s disease were randomized to follow either a carbohydrate-rich or protein-rich diet for 3 days before testing; patients on the carbohydrate-rich diet improved their maximal work capacity and exercise tolerance to submaximal workloads [Andersen and Vissing, 2008]. Aerobic training of four McArdle’s patients improved peak cycle exercise capacity, circulatory capacity, and oxygen uptake [Haller et al., 1998b].
Vitamin B6 is another potential therapeutic aid, as overall body stores of pyridoxal phosphate are depleted in McArdle’s disease due to the lack of enzyme protein to which pyridoxal phosphate is bound [Haller et al., 1983]. One patient has been shown to have a beneficial effect with vitamin B6 supplementation [Phoenix et al., 1998], but further studies need to be done. In addition, oral creatine monohydrate supplementation in a placebo-controlled crossover trial involving nine patients has been shown to alleviate symptoms and increase their capacity for ischemic, isometric forearm exercise [Vorgerd et al., 2000]. Another strategy has been the ingestion of sucrose prior to exercise to increase the availability of glucose [Vissing and Haller, 2003]. In a single-blind, randomized, placebo-controlled crossover study, 12 patients were studied. Ingestion of sucrose prior to exercise improved exercise tolerance and sense of well-being. In another comparative study, ingestion of 37 g of sucrose 5 minutes before exercise had a marked and prolonged effect on exercise tolerance in patients with McArdle’s disease. This was a more sustained effect than that seen with 75 g of sucrose 40 minutes before exercise, probably related to more continuous glucose uptake from the intestine and correspondingly higher circulating glucose levels later during exercise [Andersen et al., 2008].
Finally, a first-generation adenoviral recombinant containing the full-length human myophosphorylase complementary DNA (cDNA) has been efficiently transduced into phosphorylase-deficient sheep and human myoblasts, resulting in restoration of phosphorylase activity [Pari et al., 1999].
Debrancher Deficiency
Clinical features
Debrancher deficiency (glycogenosis type III, GSD type III) is characterized by childhood-onset liver dysfunction with hepatomegaly, growth failure, fasting hypoglycemia, and, infrequently, hypoglycemic seizures. Spontaneous resolution may occur around puberty with normal adult liver function [Smit et al., 1990], although cirrhosis and liver failure may develop later [Fellows et al., 1983]. The myopathy tends to present late in the third or fourth decade in 70 percent of cases and involves primarily distal leg and intrinsic hand muscles. It is slowly progressive and rarely incapacitating. Childhood onset has been documented in 7 of 22 patients. Two patients had exercise intolerance, cramps, and premature fatigue [Murase et al., 1973; Ozand et al., 1967], and 5 others had diffuse weakness and wasting, growth failure, and gross motor delay [Badurska et al., 1970; Slonim et al., 1982]. Myoglobinuria has been reported in one case [Brown, 1986]. Enzymatic deficiency was documented in the muscle of 16 patients, 11 of whom had no weakness [Moses et al., 1986]. However, given the young age of these patients (2–27 years), clinical myopathy may have developed later. Peripheral neuropathy has also been documented [Moses et al., 1986]. Although clinical cardiomyopathy is infrequent, cardiac involvement has been demonstrated in most patients with myopathy [Brunberg et al., 1971; Miller et al., 1972; Moses et al., 1989]. Of interest, Cleary et al. [2002] reported consistent facial features in seven patients with GSD type III, which included midfacial hypoplasia with a depressed nasal bridge and a broad upturned nasal tip, indistinct philtral pillars, and bow-shaped lips with a thin vermillion border. In addition, younger patients had deep-set eyes. Several children had clinical problems, such as persistent otitis media or recurrent sinusitis.
Laboratory data
Glucagon or epinephrine in the fasting state does not increase blood glucose, which indicates liver involvement [DiMauro and Tsujino, 1994]. All patients with myopathy have an increase in serum CK. The EMG is myopathic but also may demonstrate fibrillation activity, and the nerve conduction velocities frequently are decreased [Brunberg et al., 1971; Moses et al., 1986]. Evidence of left ventricular or biventricular hypertrophy may be seen on EKG and echocardiography [Brunberg et al., 1971; Moses et al., 1989; Smit et al., 1990].
Inheritance
There is a male predominance. The gene has been assigned to chromosome 1p21 [Yang-Feng et al., 1992]. Prenatal diagnosis can be made by a qualitative assay based on the persistence of the abnormal polysaccharide in cultured amniocytes exposed to a glucose-free medium [Yang et al., 1990].
Pathology
A severe vacuolar myopathy, which contains PAS-positive material that is digested by diastase, is seen on muscle biopsy. This corresponds to large pools of free and apparently normal glycogen particles by electron microscopy [DiMauro and Tsujino, 1994]. Excessive glycogen is also seen in skin biopsies [Sancho et al., 1990], cultured muscle [Miranda et al., 1981], endomyocardial biopsies [Olson et al., 1984], intramuscular nerves [Powell et al., 1985], Schwann cells and axons of sural nerve [Ugawa et al., 1986], and brain [Hug and Schubert, 1966].
Biochemistry
The debrancher enzyme is a single 160-kDa polypeptide with two distinct catalytic functions. After digestion of glycogen to four glucosyl units by PPL, the residual stubs are removed by the debrancher enzyme in two steps – a transferase and a glucosidase – which are located in different domains of the polypeptide [Bates et al., 1975]. Based on enzymatic and immunologic assays, debrancher deficiency has been classified into three groups [Chen et al., 1987; Ding et al., 1990]:
Molecular genetics
Human debrancher cDNA has been isolated and sequenced [Yang et al., 1992]. A number of mutations have been described in GSD type IIIa [Okubo et al., 1996, 1999, 2000; Parvari et al., 1998; Shen et al., 1996, 1997; Shaiu et al., 2000], as well as in GSD type IIIb [Shen et al., 1996]. It is interesting to note that most patients with the IIIb variant have mutations in exon 3 of the debrancher gene, which are expected to result in truncated proteins [Shen et al., 1996]. Shaiu et al. [2000] reported two frequent mutations, each of which was found in homozygous state in multiple patients, and each of which was associated with a subset of clinical phenotype in those patients with that mutation. The first mutation was a novel point mutation of a single T deletion at cDNA position 3964 (3964 delT), which was first detected in an African American patient who had a severe phenotype and early onset of clinical symptoms. The second mutation was an A to G transition at position −12 upstream of the 3′ splice site of intron 32 (IVS32-12A>G) which was confirmed in a Caucasian GSD IIIa patient presenting with mild clinical symptoms. These two mutations together accounted for more than 12 percent of the molecular defects in the GSD type III patients tested at that time.
Whereas the overall incidence of GSD type III in the United States is about 1 in 100,000 live births, it is unusually frequent among North African Jews in Israel (prevalence of 1 in 5400, carrier prevalence 1 in 35) [Parvari et al., 1997]. Another ethnic group with a high prevalence of GSD type III (1 in 3600 with a carrier frequency of 1 in 30) are individuals from the Faroe Islands [Santer et al., 2001]. The population of 45,000 of this small archipelago in the North Atlantic has its roots in colonization by the Norwegians in the 8th century and throughout the Viking age. It was concluded that, due to a founder effect, the Faroe Islands have the highest prevalence of GSD type IIIa worldwide.
Treatment
Fasting hypoglycemia should be avoided in infants and young children with debrancher deficiency through the implementation of frequent feedings and nocturnal gastric glucose infusions and uncooked cornstarch [Fernandes, 1990]. A marked improvement was noted in a 7-year-old child with severe weakness and wasting, following institution of a high-protein diet overnight for 6 months [Slonim et al., 1982]. However, a 6-month high-protein diet had no significant effect in an adult with myopathy and distal wasting [DiMauro and Tsujino, 1994].
Phosphofructokinase Deficiency
Clinical features
Muscle PFK deficiency (Tarui’s disease) (glycogenosis type VII) has been reported in more than 100 patients [Toscano and Musumeci, 2007]. Exercise intolerance with cramps and compensated hemolysis is the main clinical feature, often associated with hyperuricemia. Fixed weakness of late adult onset was prominent in three patients. Myoglobinuria has been documented in 10 of 25 patients. Six children have been reported with early severe myopathy, with or without contractures [Amit et al., 1992; DiMauro and Tsujino, 1994]. One boy also had a cardiomyopathy and died at 21 months of age [Amit et al., 1992]. None of the infantile cases had hemolysis, suggesting different molecular etiologies. Other associated multisystem signs in infants or very young children have included seizures, cortical blindness, corneal opacifications, and cardiomyopathy [DiMauro and Lamperti, 2001; Al-Hassnan et al., 2007]. Four patients had hemolysis without myopathy, and two had normal or only mildly decreased muscle activity and about half-normal erythrocyte PFK activity. This was attributed to instability of the muscle-specific subunit of PFK [Etiemble and Kahn, 1976; Kahn et al., 1975]. Minor clinical differences from McArdle’s disease include more common reports of nausea and vomiting during exercise-induced crises of myalgia, cramps, weakness, and lower frequency of attacks of myoglobinuria [DiMauro and Lamperti, 2001]. As PFK deficiency blocks the metabolism of both muscle glycogen and blood glucose, Haller and Vissing [2004] studied 5 individuals with muscle PFK deficiency and 29 patients with McArdle’s disease during continuous cycle exercise; they found that no PFK-deficient patient developed a spontaneous second-wind phenomenon under conditions that consistently produced one in patients with McArdle’s disease, and concluded that the ability to metabolize blood glucose was critical to the development of a typical spontaneous second wind.
Laboratory data
The serum CK level is usually increased in patients with muscle disease. Hemolysis is indicated by an increased serum bilirubin and moderate reticulocytosis [DiMauro and Tsujino, 1994]. Most patients have an increase in uric acid. EMG may be normal or myopathic. 31P-NMR spectroscopy shows a different pattern from that obtained in McArdle’s disease because, in PFK deficiency, as well as in other defects of distal glycolysis, glycolytic intermediates accumulate in muscle as phosphorylated monoesters and can be detected as a distinct spectral peak [Argov and Bank, 1991].
Inheritance
The gene encoding the muscle (M) subunit is located on chromosome 12 [Howard et al., 1996].
Muscle biopsy
Diagnosis can be made by a negative histochemical PFK stain [Bonilla and Schotland, 1970]. The definitive diagnosis comes from the biochemical analysis, but only if the muscle specimen is snap-frozen at the time of biopsy, as PFK is highly labile and its activity declines rapidly in specimens kept at room temperature or on wet ice; this is why partial PFK deficiencies with residual activities above 10 or 20 percent of normal should be considered skeptically [DiMauro and Lamperti, 2001]. An important differentiating feature from McArdle’s disease is the presence of an abnormal polysaccharide in some cases, particularly older patients [Hays et al., 1981]. This abnormal polysaccharide has the characteristics of polyglucosan and stains intensely with the PAS reaction, but is resistant to diastase digestion; ultrastructurally, it is composed of finely granular and filamentous material, similar to the storage material seen in branching enzyme deficiency and in Lafora’s disease [DiMauro and Lamperti, 2001].
Biochemistry
PFK (ATP:d-fructose 1-phosphotransferase) is a tetrameric enzyme under the control of three structural loci that encode three distinct subunits: namely, M (muscle), L (liver), and P (platelet) [Vora, 1983; Vora et al., 1983a]. The three subunits show variable expression in different tissues. Mature human muscle expresses only the M subunit, whereas erythrocytes express both the M and L subunits, thereby containing five isozymes. In the typical form of PFK deficiency, genetic defects of the M subunit cause total lack of activity in muscle and partial enzyme deficiency in erythrocytes, where the homotetramer L4 is responsible for the residual activity. Other tissues, such as liver, that express predominantly the L subunit are not affected.
Pathophysiology
The functional consequences of PFK deficiency are similar to those observed in McArdle’s disease and are related to the inability of muscle to generate pyruvate [DiMauro and Tsujino, 1994]. With the ischemic exercise test, venous lactate fails to rise [Tarui et al., 1965]. However, PFK deficiency differs significantly from PPL deficiency because of the inability of PFK-deficient muscle to use glucose. Due to this inability, maximal oxygen uptake has been obtained by increasing FFA availability [Haller and Lewis, 1991]. Exercise intolerance is not decreased by glucose administration, which is potentially harmful because it reduces the concentration of FFAs and ketones. This explains why high-carbohydrate meals exacerbate exercise intolerance in patients with PFK deficiency, a situation referred to as the “out-of-wind” phenomenon [Haller and Lewis, 1991]. The excessive exercise-induced degradation of muscle purine nucleotides likely accounts for the hyperuricemia seen in PFK, PPL, and debrancher-deficient patients [Mineo et al., 1987]. Finally, in PFK deficiency, as in McArdle’s disease, exercise triggers increases in heart rate, cardiac output, and blood flow, which are exaggerated relative to the muscle capacity to use oxygen [Lewis et al., 1991].
Molecular genetics
The genes encoding the subunits M, P, and L have been assigned to chromosomes 12, 10, and 21 (see Table 94-1) [Van Keuren et al., 1986; Vora et al., 1982, 1983b]. The M subunit cDNA has been isolated and sequenced [Nakajima et al., 1987], and evidence for tissue-specific alternative mRNA splicing has been documented [Nakajima et al., 1990]. At least 20 mutations responsible for muscle PFK deficiency have now been identified [DiMauro et al., 1995; Sherman et al., 1994; Tsujino et al., 1994d; DiMauro and Lamperti, 2001]. Raben and Sherman [1995] tabulated 15 disease-inducing mutations of the muscle PFK gene and several polymorphisms. These included splicing defects, frameshifts, and missense mutations in patients from six different ethnic backgrounds, supporting genetic heterogeneity of the disease. This disorder appeared to be particularly prevalent among individuals of Ashkenazi Jewish descent. The most frequent mutation in this group was an exon 5 splicing defect that accounted for approximately 68 percent of mutant alleles in the Ashkenazim. Patients with muscle PFK deficiency diagnosed in the United States have been of Ashkenazi Jewish origin, and the several mutations identified in them differ from those described in the non-Ashkenazi Italian or Japanese patients [DiMauro et al., 1995].
Treatment
Glucose is not an alternative substrate in PFK deficiency. A high-protein diet may be beneficial, although this has not been tried [DiMauro and Tsujino, 1994]. A 2-year old boy with the severe infantile form of PFK deficiency, including arthrogryposis multiplex congenita, respiratory insufficiency, slowed motor nerve conduction velocities, and abnormal electroencephalogram (EEG), had a significant improvement in strength, EMG features, and EEG pattern on the ketogenic diet [Swoboda et al., 1997]. Unfortunately, the child died of complications of pneumonia at 35 months.
Phosphoglycerate Kinase Deficiency
Clinical features
PGK deficiency (glycogenosis type IX) presents with nonspherocytic hemolytic anemia and dysfunction of the central nervous system [Boivin et al., 1974; Valentine et al., 1960]. Neurologic problems have included behavioral abnormalities, mental retardation, seizures, and strokes. In hemizygous males, the severe hemolytic anemia manifests soon after birth with jaundice, splenomegaly, and hemoglobinuria. Childhood mortality has been reported in four members of one family [Valentine et al., 1960]. Heterozygous females may be normal or may have mild chronic hemolytic anemia. Myopathic features have included myopathy in males [DiMauro et al., 1981a, 1983a; Rosa et al., 1982; Tonin et al., 1993; Morimoto et al., 2003], with exercise intolerance, cramps, and myoglobinuria. Twenty PGK variants with reduced PGK activity have been identified; myopathy was present in eight of these variants [Tsujino et al., 1995b]. Spiegel et al. [2009] reported an 18-year-old man of Arab Bedouin descent with onset of muscle cramps and recurrent myoglobinuria at age 7 years, having elevated CK, without hemolytic anemia or brain dysfunction, in whom PGK deficiency was documented in muscle and erythrocytes due to a novel mutation, T378P. This was the ninth case presenting with isolated myopathy, whereas most other patients show nonspherocytic hemolytic anemia alone or associated with brain dysfunction and a few patients have myopathy and brain involvement.
Inheritance
PGK is located on the long arm of the X chromosome (see Table 94-1). The defect is expressed in fibroblasts, and prenatal diagnosis is possible [DiMauro and Tsujino, 1994].
Muscle biopsy
PAS stain for glycogen was mildly increased in one patient. Electron microscopy demonstrated glycogen accumulation in all patients [DiMauro and Tsujino, 1994].
Biochemistry
Human PGK is a single polypeptide encoded on the X chromosome for all tissues except spermatogenic cells [DiMauro and Tsujino, 1994]. The complete amino-acid sequence of normal human PGK has been determined [Huang et al., 1980a, 1980b], and mutations have been identified. Patients with hemoglobinuria and those with myopathy demonstrate genetic heterogeneity in enzyme activities. PGK mutants associated with myopathy have been named Creteil [Rosa et al., 1982], New Jersey [DiMauro et al., 1983a], Alberta [Tonin et al., 1993], Hamamatsu [Sugie et al., 1989, 1998], Shizuoka [Fujii et al., 1992], and North Carolina [Tsujino et al., 1994e]. As PGK is a monomer, there are no tissue-specific isoforms, with the exception of PGK2, which is confined to spermatogenic cells; therefore, the defect should be expressed in all tissues. In three patients with myopathy, PGK was significantly decreased in erythrocytes, leukocytes, platelets, fibroblasts, and muscle culture [DiMauro et al., 1983a; Rosa et al., 1982; Tonin et al., 1993].

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


