Convulsive Status Epilepticus
Frank W. Drislane
Susan T. Herman
Peter W. Kaplan
HISTORY
Status epilepticus (SE) may have been described as early as 600 to 700 BC in cuneiform writings, one of which notes: “If the possessing demon possesses him many times during the middle watch of the night, and at the time of his possession his hands and feet are cold, he is much darkened, keeps opening and shutting his mouth, is brown and yellow as to the eye. It may go on for some time, but he will die” (1,2). Caelius Aurelianus, a scholar from the late Roman empire, noted that “fits can recur … even in the same day” (3).
In the 17th century England, Thomas Willis wrote,” … when as fits are often repeated, and every time grow more cruel, the animal function is quickly debilitated … [and] …. the vital function is by little and little enervated, till at length, the whole body languishing, and the pulse loosened, and at length ceasing, at last the vital flame is extinguished” (4).
Reports of SE are few through the 18th century, but in the 1800s, the study of epilepsy flourished in Paris at the Salpêtrière that, with 8000 patients, was Europe’s largest asylum. Physicians there, including Charcot, provided extensive clinical descriptions of SE. The topic was brought to modern attention in the university dissertation of Calmeil in Paris, who reported that the phrase derives from the patients’ use of the term: “C’est ce que les malades appellent entre eux l’état de mal” (5). [“It’s what the patients among themselves called a bad condition (of epilepsy).”] He detailed the illness’s severity and a sequence of seizures without pause that forebode a poor outcome. The use in (Latinized) English of SE appeared when Bazire translated the lectures of Trousseau, who wrote, “You, however, have heard of circumstances where fits have lasted two or three days, and ended in death. It is in these cases that one spoke of, at the Salpêtrière or at Bicêtre, of an état de mal (status epilepticus)” (6). Rather than simply repeated seizures, SE was then assumed to be a distinct entity representing the “maximum expression of epilepsy with its own particular characteristics, although restricted at the time to tonic-clonic seizures” (1).
In 1903, Clark and Prout described the clinical and pathological appearance of SE in 38 patients, detailing a fusion of successive convulsions over 2 to 9 days, to the point of coma and exhaustion, along with changes in respiration, temperature, and pulse (7). The presentation was “… composed of delirium, stupor or coma …. and a variety of psychic states, which have for their basis cortical discharges.” They listed three stages of SE, with the third described as stupor—likely corresponding to the nonconvulsive SE that follows generalized convulsive status epilepticus (GCSE) with “electromechanical dissociation” and continued electrochemical seizure activity in the brain after cessation of clinically observable convulsions, sometimes now referred to as “subtle” SE (8).
Relatively little progress in the management of SE was made until the 20th century. The first modern meetings on SE, the Marseille Colloquia of 1962 and 1964, promulgated better definitions and classification for seizures and SE and suggested that SE was the prolongation of many different types of epileptic seizures. Gastaut, the organizer of the meetings, stated that there were “as many forms of SE as there were types of seizures.” Today, most literature recognizes SE’s own particular pathophysiology, not always identical to that of prolongation of an individual seizure (9).
Convulsive SE can be characterized clinically without electrophysiologic data, but Berger’s development of the electroencephalography (EEG) in 1929 led to valuable understanding of the different forms of SE, especially for nonconvulsive SE (10) (see Chapter 29). Since the 1960s, increasing scientific and clinical attention has turned to the physiologic underpinnings, neurochemistry, and pharmacology of SE, most recently with genetic investigations and functional imaging.
DEFINITIONS
Gastaut defined SE as “an epileptic seizure that is so frequently repeated or so prolonged as to create a fixed and lasting epileptic condition” (11). No duration was specified. The International League against Epilepsy specified “a single epileptic seizure of >30-min duration or a series of epileptic seizures during which function is not regained between ictal events in a 30-minute period” (12). Although without a definite scientific basis, 30 minutes has been the standard criterion for duration in most clinical studies (13).
SE is often a series of individual seizures, without recovery between seizures. Alternatively, a single convulsion can be very prolonged, and many convulsive seizures progress to become nonconvulsive SE, in which the EEG can demonstrate ongoing (nonconvulsive) seizure activity. If recovery occurs between individual convulsions, this is considered a clustering of seizures rather than SE.
While experimental studies on SE show evidence of neurologic damage after 30 minutes, a shorter duration of seizures is currently felt to warrant urgent treatment. Prospective clinical trials must define SE as diagnosable expeditiously, and urgent treatment is a clinical necessity. Thus, the largest trial of different antiepileptic drugs (AEDs) for the treatment of GCSE (14) considered it mandatory to begin treatment (and make the
diagnosis) within 10 minutes. Later, an “operational” definition of GCSE proposed that 5 minutes was the time by which SE should be interrupted to avoid morbidity, mortality, or refractory SE (15). The duration considered to characterize SE has been (just barely) agreed upon for GCSE; it is reasonable to use 30 minutes as a clinical definition for other types of SE.
diagnosis) within 10 minutes. Later, an “operational” definition of GCSE proposed that 5 minutes was the time by which SE should be interrupted to avoid morbidity, mortality, or refractory SE (15). The duration considered to characterize SE has been (just barely) agreed upon for GCSE; it is reasonable to use 30 minutes as a clinical definition for other types of SE.
PATHOPHYSIOLOGY OF STATUS EPILEPTICUS
Episodes of SE appear clinically to begin in the same way as individual seizures of the same type, but there are many different types of seizures and SE, and it is rare to capture the beginning of SE on an individual EEG. Following initiation, almost half of seizures lasting from 10 to 29 minutes cease spontaneously, without treatment (16), and most individual secondarily generalized convulsions cease on their own in under 2 minutes and the patient recovers thereafter (17). Some seizures, however, continue despite the brain’s intrinsic capacity to terminate shorter seizures, and they progress to the longer episodes of SE.
It is unclear exactly what facilitates the transition from individual seizures to seemingly self-sustaining prolonged seizures or SE, but this transition appears to depend substantially on the relative balance of excitatory and inhibitory electrochemical function at the time—at intracellular, membrane, extracellular, and interneuronal levels. To a large extent, SE implies a failure of the typical inhibitory function of the brain in terminating seizures as usual (18).
There are probably several pathophysiologic mechanisms that facilitate the continuation of seizures, and in some seizures, the balance of electrochemical function appears to shift, unfavorably, toward excessive excitatory activity as seizures progress. In experimental animals, one of the primary cellular changes in the transition from prolonged seizures to refractory SE is a marked and progressive impairment of gamma-amino butyric acid (GABA) agonist-mediated inhibitory function, in turn favoring recurrent excitatory circuits (19). During seizures, neurotransmitter receptor changes occur almost immediately, with gene transcription and new peptide production after several minutes. GABA receptor modification occurs within 45 minutes, showing rapid functional plasticity (20). The electrochemical activity of ongoing seizures also induces gene transcription leading to the production of numerous molecular factors with excitatory or inhibiting effects on continuing seizures. Among the many are galanin (which inhibits seizures and SE) (21), substance P, neuropeptide Y, somatostatin, and dynorphin.
Early in SE, there is a marked and maladaptive diminution in the number of GABA-A receptors on neuronal surfaces (22), in large part due to progressive internalization or endocytosis of those receptors (23). This likely contributes to the reduced effectiveness of GABA agonists in terminating seizures. As seizures progress, in both experimental animals and in human studies, they appear to become resistant to many AEDs (24), especially those acting through the GABA system, such as benzodiazepines (BDZs) and barbiturates (20), and those drugs become less useful in treating SE.
As SE continues (concomitant with the decay in GABA receptor function), there is increasing excitatory activity of glutamate receptors, especially the N-methyl-D-aspartate (NMDA) receptor, promoting and prolonging the hyperexcitable state (18,25,26). When glutamate binds to the NMDA receptor, membrane ion flow can still be blocked by magnesium, but with sufficient repetitive depolarization (as occurs during seizures), the magnesium is released and the ion channel opens more completely in a “use-dependent” facilitation of glutamatergic activity (27). NMDA receptor antagonists are not effective in blocking the initiation of SE, but as seizures continue, with repetitive depolarization, they appear to become more effective anticonvulsants in the same “use-dependent” manner (28). At that point, glutamate antagonists (such as ketamine) may become more effective treatments (29).
As seizures continue, stimulation of glutamate (especially NMDA) receptors leads to excessive sodium and calcium entry into neurons, and this may cause cell death. Gene activation during SE may lead to the production of kinases and phosphatases that accelerate the harmful process. Later, inflammation, glial cell changes, and neuronal sprouting or cell loss may contribute to the refractoriness of subsequent seizures (18). The potential neuronal injury from prolonged seizures or SE may depend somewhat on the intensity (or frequency) of the epileptiform discharges (30).
There is abundant experimental (if much more modest clinical) evidence that neuronal damage results from GCSE, particularly when prolonged. A century ago, this damage was attributed to ischemia, but in the 1970s, Meldrum and colleagues showed that excessive excitatory neuronal activity was the cause of nerve cell damage, at least in experimental animals with prolonged GCSE (31, 32 and 33). In baboons given bicuculline to induce SE, prolonged convulsions with continued EEG spike discharges for 1 to 5 hours caused neuronal damage in the neocortex, cerebellum, and hippocampus (31, 32, 33 and 34). By “clamping” blood flow, brain oxygenation, and glucose supply to preserve homeostasis during SE, they showed that hypoperfusion was not crucial in the development of such damage. Neuronal injury still occurred in the hippocampus, and it appeared to be due to excitatory neurotransmitter activity. Thus, the prolonged ongoing cellular electrochemical activity of SE itself (rather than concomitant physiologic derangements) was sufficient to cause neuronal injury, although systemic abnormalities also contributed to this injury. This hippocampal damage may correlate with prolonged memory and other cognitive dysfunction in humans following SE.
It is very difficult to prove that SE leads to neuronal damage in humans (as it clearly does in experimental animals exposed to prolonged, intense electrical stimulation), in part because SE is seldom fatal without causation by other severe neurologic illnesses that themselves cause neuropathologic damage independent of the SE. Nevertheless, a disproportionately decreased neuronal cell density was seen in the hippocampus of some patients who died from GCSE (but not NCSE) (35). Forms of SE other than GCSE may not be as harmful and may not require exactly the same treatment, but concern for the possibility of such damage leads to a clear mandate for urgent treatment.
EPIDEMIOLOGY AND CAUSES
Studies of the incidence of SE vary tremendously by definitions and different populations studied. The corrected, age-adjusted incidence of SE was 17.1 cases per 100,000 population per year in Germany and 54.5/100,000 in subjects greater than 60 years old; the case fatality rate was 9.3% (36). This study included all forms of SE. Most were due to remote symptomatic causes such as cerebrovascular disease; the mean age of adult patients was 65 years. SE is more common in children under 1 year and in the elderly. The incidence of SE is several times that in developing countries, particularly in children (37).
The incidence of all forms of SE was 41/100,000 in Richmond, Virginia, but 20/100,000 in the white population, similar to that in Germany (38). The incidence was higher in African Americans and also likely higher because of ascertainment in an urban medical setting. Extrapolation from the same database suggests an incidence of up to 50/100,000 per year, substantially higher than most estimates (13). In another study, mortality of SE was 19% within the first 30 days, highly associated with the acute cause of SE (39). Short-term case fatality is generally 20% to 25% when considering GCSE alone, and in all series it is primarily due to the underlying illness causing SE (13). In another study of patients with afebrile SE, 32% had recurrence of SE within 10 years of follow-up (40). Recurrence is higher in patients with progressive neurologic disorders.
Causes of SE are extremely numerous. They may be grouped into larger categories similar to the causes of seizures and epilepsy syndromes (12): “symptomatic,” that is, a consequence of a known or suspected cerebral dysfunction such as head injury, vascular disease, infection, tumor or other mass lesion, toxic or metabolic disturbances, or following surgery; “idiopathic,” such as in the idiopathic generalized epilepsies (IGEs), almost always related to a known or presumed genetic-based syndrome; or “cryptogenic,” in which there is a presumed underlying cause, but it cannot be identified. The IGEs lead to SE relatively infrequently, and the SE is usually nonconvulsive (see Chapter 29), except for the unusual case of generalized convulsions upon awakening (4).
The most common cause of SE in most larger series, especially in nonhospitalized patients, is “remote symptomatic” (with the cause present for more than a week), often due to earlier injuries such as stroke and often with more immediate exacerbation by some systemic problem such as infection, medications, or AED withdrawal (24,42). The next largest category of causes is probably an exacerbation of earlier epilepsy, also commonly with an acute precipitant such as lowered AED levels, infection, or metabolic disturbance. In acute care hospitals, most SE patients have “acute symptomatic” causes, that is, due to a recent (within a week) neurologic injury or illness, the most common example of which is a new stroke (43). Most have no history of seizures and are often older adults with acute and severe medical or neurologic illnesses.
In order to discuss diagnosis, management, and outcome, it is important to recognize the tremendous variety of clinical situations that fall under the rubric of SE and to specify exactly which clinical syndrome a patient has. SE, like seizures, can be organized clinically into those with a focal onset and those with a generalized onset—although many generalized cases cannot be divided easily into those of a primarily generalized nature versus those with a focal onset and secondary generalization (Table 28.1). Most epidemiologic studies do not specify the type of SE, but those that do indicate that GCSE is the most common and accounts for about half of all SE. Forms of SE with prominent motor signs, whether convulsive or myoclonic, are covered in this chapter; those with primarily nonmotor manifestations are described in the Chapter 29.
Table 28.1 Different Forms of Status Epilepticus | |||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||
GENERALIZED CONVULSIVE STATUS EPILEPTICUS
Presentation, Clinical Observations
GCSE is the best described form of SE and has the greatest morbidity, mortality, and clinical urgency for treatment (44). It may begin as primary generalized convulsive seizures or evolve from
a focal or complex partial seizure and progress to generalized convulsions. Most GCSE has some evidence of a focal onset or focal lesion (13), that is, about 70% to 80% is secondarily generalized (42). Some patients have recurrent seizures without recovery of consciousness between seizures; others evolve from the first convulsion into a more prolonged seizure, with motor manifestations becoming less regular clonic movements and regular or irregular myoclonic jerking activity, in a continuous epileptic (“ictal”) state (44). Primary GCSE is relatively uncommon, often occurring as a result of AED discontinuation or the consequences of inappropriate AEDs or other drugs, or systemic illness in patients with IGEs (41).
a focal or complex partial seizure and progress to generalized convulsions. Most GCSE has some evidence of a focal onset or focal lesion (13), that is, about 70% to 80% is secondarily generalized (42). Some patients have recurrent seizures without recovery of consciousness between seizures; others evolve from the first convulsion into a more prolonged seizure, with motor manifestations becoming less regular clonic movements and regular or irregular myoclonic jerking activity, in a continuous epileptic (“ictal”) state (44). Primary GCSE is relatively uncommon, often occurring as a result of AED discontinuation or the consequences of inappropriate AEDs or other drugs, or systemic illness in patients with IGEs (41).
Clinically, GCSE is readily recognizable by its motor manifestations, which are often dramatic. Typically, convulsive activity consists of paroxysmal or continuous bilateral, usually rhythmic, tonic movements or clonic shaking of the limbs, commonly with facial and truncal manifestations as well, and with loss of consciousness. Convulsions may have some lateralized emphasis, even though the physiologic seizure activity is bilateral or generalized. If the generalized convulsion is primary, the patient may exhibit no movement for a second or two, followed by the tonic phase of a seizure in which muscle contraction occurs throughout the whole body, including a tightening of chest muscles giving rise to the “epileptic cry.” The tonic phase typically lasts about 10 seconds (45,46) and is interrupted by the clonic phase, with particularly rapid and forceful flexion contractions of the arms. Sometimes movements are more subtle, and there may be a faster quivering. Typically, there is a progressive lessening of the violence of the convulsions, often within the first minute of SE. Eventually the frequency of clonic contractions lessens, and they cease (45,46). The patient is then flaccid, but there may be a large generalized myoclonic jerk at the end of the motor phase. The tonic, and then clonic, phases of an individual seizure seldom last more than 2 minutes (17). As GCSE progresses, motor manifestations may lessen and include just relatively subtle eyelid or facial or other myoclonic jerking, or motor phenomena may disappear entirely (8). Progression to this “subtle” GCSE (below) is not characteristic of primary GCSE (44). Repeated seizures also diminish in duration and intensity over time (44,47).
Consciousness is lost completely at the beginning of GCSE with the first convulsion, or earlier, and the patient is apneic during the tonic and clonic phase. The eyes are usually open and may show pupillary dilation. Tachycardia, hypertension, and other autonomic changes are common. Following an isolated convulsion, muscle tone and consciousness return gradually, usually with some improvement within minutes, but in SE recovery does not occur before the next generalized convulsion or progression to more subtle or nonconvulsive SE.
THE EEG OF GENERALIZED CONVULSIVE STATUS EPILEPTICUS
EEG patterns in SE undergo evolution, usually paralleling the clinical manifestations. Generalized convulsions typically show bilaterally symmetric activity (behavioral and EEG) from the onset and may progress through a stereotyped sequence of clinical and electrographic manifestations (8,47) (Fig. 28.1). The initial brief interruption in behavior is often accompanied by widespread voltage attenuation with fast frequencies at 20 to 40 Hz, sometimes called “electrodecremental.” This may be a widespread desynchronization of the background, but with implanted EEG electrode recording, very rapid (>200 Hz) activity may be evident in the area of seizure onset (not evident in routine, surface EEG), suggesting a role for pathologic organization of networks or circuits of neurons in the generation of seizures (48). Within seconds, as the initial tonic contraction occurs, generalized rhythmic electrographic seizure activity, including rapid spike discharges, gradually increases in voltage and decreases in frequency to approximately 10 Hz.
Not all episodes of GCSE begin this way. Some start with early electrographic signs of an IGE that, within seconds, assumes the clinical and EEG picture of a tonic and then clonic seizure (Fig. 28.2A-D). Still others begin as focal-onset seizures and generalize rapidly. Within seconds, all three can appear very similar clinically and on EEG.
During the clonic phase of the seizure, there is rhythmic slow activity, sometimes with interspersed spikes, for 10 seconds or more (Fig. 28.2B). Generalized spike and slow waves or polyspike and slow-wave activity correspond to the frequency of the clinical manifestations.
Many of the initial EEG rhythms of GCSE are obscured by the muscle activity of the convulsion, unless the patient has received paralyzing drugs. It is often only when the motor manifestations lessen that the EEG becomes interpretable. Sometimes, epileptiform discharges can be seen at the vertex. The superimposed electromyographic activity, however, may exhibit its own characteristic rhythmic appearance, synchronous with the clinical jerking activity, and this muscle activity on the EEG still indicates generalized convulsions (Fig. 28.2C).
If the seizure ends (or between seizures) the discharging activity gives way to a slow, encephalopathic background in all areas, or sometimes a very suppressed background, before a gradual resumption of a normal drowsy and then waking pattern (Fig. 28.2D). In GCSE characterized by repetitive seizures with impaired consciousness, the intervening EEG shows diffuse slowing, voltage attenuation, and disorganization. Between seizures, there are often persistent generalized epileptiform discharges with a rhythmic appearance, but slower than during the convulsion (49).
As GCSE becomes prolonged, the EEG becomes more discontinuous, and clinical manifestations become more subtle, often with relatively minimal eyelid or facial or other myoclonic jerking, nystagmoid eye movements, or even absence of motor movements, all while the EEG continues to show repetitive, rhythmic epileptiform discharges. When the visible motor manifestations of GCSE cease, this can be termed “subtle” GCSE (below) (8).
Some have proposed that there is a characteristic sequence of EEG changes during GCSE, based on recordings from animals and humans during various stages of SE (Fig. 28.3) (8): (i) Seizures are initially discrete and followed by background slowing and attenuation until the next seizure begins. (ii)
The seizures merge gradually, with waxing and waning of voltage and frequency. (iii) Eventually, seizures become continuous, although ictal discharges are frequently asymmetric, reflecting the partial onset of many of the seizures. (iv) With time, seizures become discontinuous; there are brief (0.5 to 8 seconds) periods of generalized voltage attenuation or flattening. (v) In late SE, generalized periodic epileptiform discharges (GPEDs) or bursts of polyspikes with frequencies from 0.5 to 4 Hz arise from a relatively flat background. “Subtle” SE often corresponds to EEG stages 4 and 5 (8). Patients are typically comatose, with minimal or no motor manifestations at this point (8,49).
The seizures merge gradually, with waxing and waning of voltage and frequency. (iii) Eventually, seizures become continuous, although ictal discharges are frequently asymmetric, reflecting the partial onset of many of the seizures. (iv) With time, seizures become discontinuous; there are brief (0.5 to 8 seconds) periods of generalized voltage attenuation or flattening. (v) In late SE, generalized periodic epileptiform discharges (GPEDs) or bursts of polyspikes with frequencies from 0.5 to 4 Hz arise from a relatively flat background. “Subtle” SE often corresponds to EEG stages 4 and 5 (8). Patients are typically comatose, with minimal or no motor manifestations at this point (8,49).
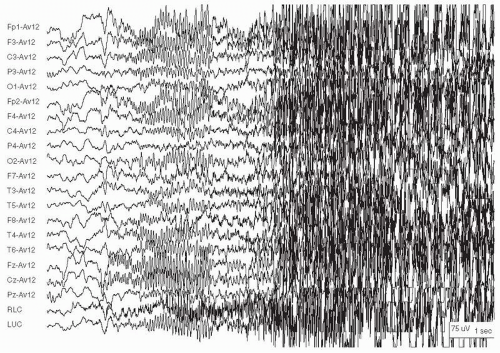 Figure 28.1 A generalized tonic-clonic seizure in a 20-year-old woman with cryptogenic generalized epilepsy. At the onset there is diffuse frontally maximal low-to-moderate voltage beta activity, followed by diffuse muscle artifact as lowamplitude clonus begins. |
Sequential EEG changes, such as the decreased prominence of ictal discharges and increase in intervening background attenuation (in parallel with the diminution of clinical manifestations), represent progressive neuronal dysfunction, possibly due to metabolic depletion (8). As the EEG “deteriorates,” motor manifestations become more subtle or disappear entirely.
Other investigators have not found the same sequence of EEG changes (50, 51 and 52). While these EEG patterns were seen frequently, they often occurred out of sequence or in only some patients, and some patients persist in one EEG pattern throughout the course of SE. Evolution of the EEG patterns depends on the etiology of SE and the baseline condition of each patient, time from SE onset, type of SE, duration of seizure activity, and treatment. Such variation makes classification of SE based purely on EEG criteria difficult and adds to the controversy regarding which patterns are “ictal.” Stage 4 above usually has subtle or no motor manifestations but has very frequent and rhythmic epileptiform discharges and is usually considered to represent NCSE. Stage 5 is far more controversial as to its ictal nature.
There is no definite difference in clinical significance if SE is manifested on EEG by recurrent discrete electrographic seizures or by continuous epileptiform discharges. In one series of focal SE, there was no significant difference in the etiologies and outcome of patients with either continuous discharges or recurrent discrete seizures on EEG (53). In another series, patients with continuous clinical seizure activity appeared to be sicker overall than those with discrete seizures (54), but the management and outcome does not differ much between the two patterns.
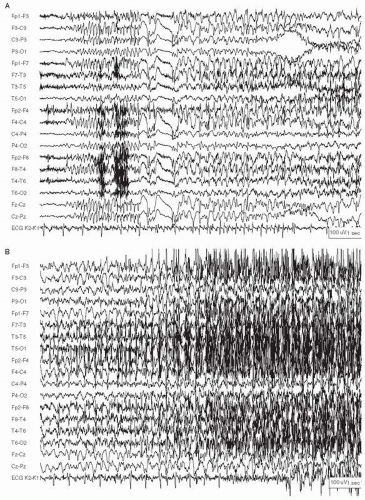 Figure 28.2 A: A generalized tonic-clonic seizure in a 43-year-old man with idiopathic generalized epilepsy and frequent episodes of status epilepticus. The seizure begins with widespread frontally maximal alpha activity, then 5-Hz spike-and slow-wave discharges. B: Continuation of the seizure in A. Slower rhythmic activity, with admixed spike and wave discharges, gradually becomes obscured by muscle activity. |
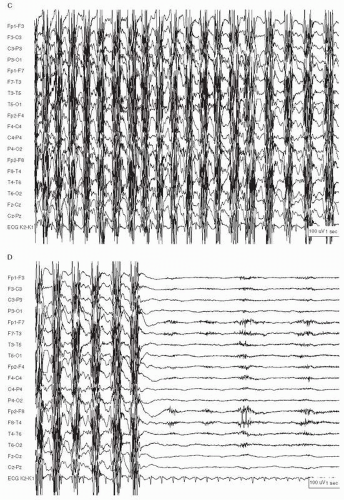 Figure 28.2 (Continued) C: After a period of EEG activity nearly entirely obscured by muscle activity (not shown here), there is evolution into generalized 1.5-Hz bursts of EMG artifact corresponding to the patient’s generalized clonic jerking. D: The seizure ends and is followed by diffuse background voltage attenuation. |
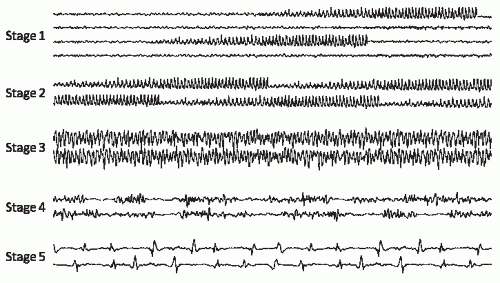 Figure 28.3 Stages of status epilepticus, illustrated by single-channel recordings showing EEG evolution (see text for details). Stage 1: Discrete seizures with intervening normal EEG. Stage 2: Merging seizures, with some intervening normal EEG. Stage 3: Continuous ictal activity. Stage 4: Discontinuous ictal activity with intervening periods of a flat background. Stage 5: Somewhat periodic epileptiform discharges on a low-voltage background. |
COMPLICATIONS, MORBIDITY
GCSE engenders major physiologic changes, several of which may contribute to its significant potential for morbidity and mortality (42). At the beginning of GCSE (as with isolated convulsions) heart rate and blood pressure increase rapidly, along with a substantial surge of catecholamines into the blood stream. Cardiac arrhythmias, including bradyarrhythmias and tachyarrhythmias, are common and sometimes serious; cardiac arrest may occur (55). Blood pressure and glucose concentrations increase early but return to baseline levels after several minutes and may even decrease further (56). Hypoperfusion can affect the liver and pancreas. Respiratory rhythm disturbances are common, as is aspiration pneumonia, often leading to fever. Prolonged treatment may be associated with infections or sepsis, thrombophlebitis, and pulmonary emboli.
Metabolic complications include electrolyte disturbances such as hyponatremia. Peripheral leukocytosis and cerebrospinal fluid (CSF) pleocytosis are common. Increased intracranial pressure and cerebral edema may develop, especially in children (57). Disseminated intravascular coagulation can lead to multiorgan failure. Fractures of long bones are common, particularly with falls and trauma, as are vertebral compression fractures. Falls also cause facial, dental, skull, and many other injuries.
Violent muscle contractions increase lactate levels, contributing to a systemic acidosis, and systemic temperatures often rise significantly (42). Prolonged muscle contraction can also lead to rhabdomyolysis, and myoglobinuria can cause renal failure. Hyperthermia may be due to continued convulsions or infection, for which the underlying cause must be sought and treated. Hyperthermia due to the SE itself usually resolves with prompt treatment of the SE.
Imaging studies, including CT, MRI, and PET scans, have shown substantial changes in brain tissue with SE, although many resolve with time, and their clinical implications are often uncertain (58). There are some well-detailed cases of permanent and severe tissue damage, such as hippocampal atrophy, reasonably attributed to very prolonged SE alone—and accompanied by neuropsychologic deficits (59).
Studies (most of which are pediatric and retrospective) have found minimal cognitive morbidity following SE. Several found negative consequences, but it is unclear whether they are due to the underlying illness or SE itself (60,61). Comprehensive neurologic and neuropsychologic evaluations before and after SE are seldom available; patients with progressive illnesses worsen, whether or not related to the SE; and neurologic deficits may fluctuate with time. It is also difficult to control for the influence of AEDs and other medications.
One group studied 193 children with SE, almost all GCSE, and concluded that “the major neurologic sequelae are usually due to the underlying insult rather than to the prolonged seizure itself” (60). Another group performed thorough neuropsychologic testing on 15 adults with prior epilepsy (without underlying lesions or symptomatic causes) before and after episodes of SE, finding no significant cognitive morbidity accruing from the SE and suggesting that the morbidity in other series comes from the underlying etiology, not from the SE itself (62). Long-term cognitive complications of SE appear to be relatively uncommon, occurring primarily in the most prolonged and severe cases.
About 10% of chronic epilepsy presents with an episode of SE (63). Conversely, about 30% to 40% of survivors of GCSE develop subsequent epilepsy (60,64,65), four times the rate following a single acute symptomatic seizure. SE might lead to epilepsy, but alternatively, patients more likely to have later epilepsy may present with more severe seizures at the onset, that is, SE may represent a marker for a more severe epileptic process that has already begun (66). While chronic epilepsy may follow SE and SE is a risk factor for such epilepsy (67), it does not appear to be causal in most.
OUTCOME, MORTALITY
Mortality in “the status epileptici” (with all forms taken together) is approximately 20% to 25% (13) with most studies consisting primarily of GCSE. Even for GCSE, outcome varies considerably and depends heavily on etiology and possibly on other factors. In one of the largest prospective, population-based studies of SE,
case fatality was 3% for children and 26% for adults—using a 30-minute definition and including all types of SE (13). Almost all studies conclude that most of the morbidity and mortality of SE derives from the illness precipitating the SE rather than from the SE itself (68,69). By one estimate, no more than 2% of fatalities comes from the SE itself (70).
case fatality was 3% for children and 26% for adults—using a 30-minute definition and including all types of SE (13). Almost all studies conclude that most of the morbidity and mortality of SE derives from the illness precipitating the SE rather than from the SE itself (68,69). By one estimate, no more than 2% of fatalities comes from the SE itself (70).
Etiology is overwhelmingly the most important prognostic factor. Short-term mortality is highest when SE occurs in the setting of an acute insult, especially stroke and anoxia (13,39,65,71). Patients with multiple medical problems, including sepsis, fare poorly, while those with SE due to tumors, trauma, alcohol abuse, or other drugs have an intermediate or lower mortality (13,42). Generally, the most favorable etiology is epilepsy itself with some exacerbating factor (reduced AEDs, fever, sleep deprivation, or an intercurrent illness or other precipitant) prompting the SE. Older patients with a symptomatic cause of GCSE have a mortality of >50% (43). Presentation in coma (vs. stupor or confusion) confers a poor prognosis (72), but this may also be related to the severity of the etiology.
Children with SE have a low mortality but the elderly a much higher one. In one series, SE patients over age 60 had a mortality of nearly 40% and those over 80 nearly 60% (73). Most SE types in the elderly are focal, or focal with secondary generalization (totaling 74%; (73)), reflecting the focal pathology, particularly stroke—the most common cause of SE in the elderly. Younger patients with earlier epilepsy had a much lower mortality (74). Mortality of SE increases with age, but this may be explained largely by the increasing incidence of acute medical illness, such as strokes and other concomitant conditions (68,75).
The duration of SE may influence outcome. Experimentally induced seizures in rodents become more refractory to treatment with BDZs after 45 minutes (compared to those lasting 10 minutes) (19). In clinical studies (not always controlled for etiology) patients with 2 or 4 hours of SE had more morbidity than those with shorter episodes (24,76). In the largest epidemiologic study of duration and SE, patients whose SE lasted an hour or more had a 32% mortality within 1 month versus 2.7% for shorter durations, even corrected for other factors by multivariate analysis (77), but there was no definite correlation between duration and mortality beyond 1 hour. When controlled for etiology and other factors, duration of SE was not a prognostic factor in at least two studies (39,78), indicating that prolonged SE should not be considered untreatable—unless the etiology is particularly ominous. Patients with shorter SE durations do better, at least in large part, because the SE has more benign causes. It has been impossible to separate duration (beyond the first hour) conclusively from the influence of etiology.
Appropriate treatment of SE appears to reduce morbidity and mortality, but it is impossible to carry out a randomized trial of treatment and nontreatment. A study comparing two centers found that poorer quality treatment (primarily the use of inadequate BDZ doses early in SE) led to worsened outcome (79). Another found that most treatment was inadequate and correspondingly ineffective, also suggesting that adequate doses of appropriate AEDs led to better outcomes (80).
The mortality of SE may be declining over the past few decades—often attributed to faster recognition and treatment (81). It is possible, however, that a wider variety of SE types is now being diagnosed, including more benign cases with lower morbidity and mortality, thus increasing the reported incidence of SE and improving the reported outcome. One study reported a recent decrease in mortality, at least when myoclonic SE (usually following cardiac arrest and anoxia) was excluded (81).
TREATMENT OF GENERALIZED CONVULSIVE STATUS EPILEPTICUS
Clinical management of SE is beyond the scope of a neurophysiology chapter but will be alluded to briefly. Although there is a substantial difference between the duration of SE that leads to neuronal damage in experimental animals and the shorter duration of SE now felt to warrant prompt treatment in patients, seizures should be treated before they last 30 minutes, and certainly by the time of “impending status” (18). The “operational” definition above recommended treatment within 5 minutes to avoid morbidity, mortality, or refractory SE (15). This applies primarily to GCSE.
As an emergency condition, GCSE warrants admission and intensive care unit (ICU) care in most cases, with urgent attention to airway, breathing, circulation, and basic medical care, addressing the underlying cause of SE and the medical illness causing or worsening it. There has been just one large randomized controlled trial of different AEDs for GCSE (14). Based primarily on that trial, several other studies, and common practice, treatment recommendations usually begin with sufficient doses of intravenous (i.v.) BDZs, the most rapidly acting medications (82). With its longer duration of action, lorazepam is frequently preferred (82, 83 and 84). This should almost always be followed by i.v. infusion of a longer acting AED such as phenytoin, fos-phenytoin, phenobarbital, valproate, or levetiracetam (82,84, 85 and 86). If the first agent is unsuccessful in treating SE, a second drug (often one of the other i.v. AEDs) is used frequently (87). Often adjunctive use is made of enterally administered carbamazepine, topiramate, and other AEDs unavailable in i.v. form.
In a large series, the treatment success rate in patients with overt GCSE was 56%, but only 15% with subtle SE (14). Lorazepam had the greatest success, terminating seizures in 65% of cases of overt GCSE. Phenobarbital (58.2%), diazepam followed by phenytoin (55.8%), and phenytoin alone (43.6%) followed, but these differences were not statistically significant (except for lorazepam vs. phenytoin). If SE continues after the first AED, a second AED succeeds in stopping SE less than 10% of the time (and even less with subtle or purely electrographic SE) (88). At this point, SE is considered refractory, and most experts move on to more definitive or aggressive treatment with continuous intravenous (C-IV) AEDs.
Common causes of treatment failure include inadequate dosing of the initial AEDs, failure in diagnosis of the cause or underlying etiology (which must be treated), inaccurate diagnosis with psychogenic nonepileptic seizures, and failure to maintain adequate AED therapy after the initial treatment—leading
to a relapse of SE, a common complication and often one signifying or predicting worsened outcome (89).
to a relapse of SE, a common complication and often one signifying or predicting worsened outcome (89).
REFRACTORY STATUS EPILEPTICUS
Most SE responds to appropriate use of AEDs, but over time, with continued seizures, SE may become refractory to treatment. The primary causes of refractory status epilepticus (RSE) are similar to those of SE overall, including stroke or central nervous system (CNS) tumor (20%), epilepsy-related (20%), toxic-metabolic encephalopathy (19%), CNS infection (19%), hypoxia-ischemia (12%), and traumatic brain injury (5%) (90). The minimal duration of seizure activity to define RSE varies, from unspecified to 2 hours (84,91,92). Most epileptologists use the term refractory without regard to temporal duration, but rather for SE that has not responded successfully to treatment with two AEDs (some require three), usually including one BDZ and a longer acting AED—administered promptly and adequately (75,84,91, 92 and 93).
The prevalence of RSE varies from 10% to over 30% of all SE, depending on definitions (84,90). In the VA Cooperative Study of GCSE treatment, RSE, defined as continued seizures after two AEDs, occurred in 38% of 384 patients with overt GCSE and 82% of 134 patients with subtle GCSE (14). In a retrospective study of all types of SE, RSE (defined as lasting more than 60 minutes, with failure of two AEDs) occurred in 31% of 83 episodes of SE in 74 patients (84). RSE was more common in patients with nonconvulsive SE (88% vs. 26% with other forms of SE) and focal motor SE. In children, about 40% of SE becomes RSE, defined as 60 minutes of seizures unresponsive to two AEDs (94). In one series of ICU patients (with SE identified and treated quickly) SE became refractory in 25% and was fatal in 21%; continuous SE had a higher case fatality rate than intermittent SE (95).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


