Figure 34-1 Symmetric facial weakness and skin laxity.
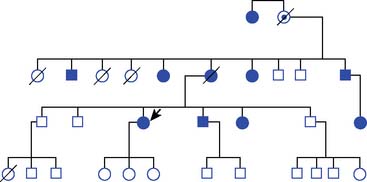
Her family history was significant for multiple similarly affected family members shown in the pedigree (Fig. 34-2). Her position in the pedigree is indicated by an arrow. Many affected members were first identified by eye complaints of “dry eyes.” Corneal lattice dystrophy was diagnosed by slit lamp examination. A maternal cousin, who was also affected, had received genetic counseling and was told she had “Finnish amyloidosis.”
Evaluation included negative or normal angiotensin-converting enzyme, serum protein electrophoresis (SPEP) with immunofixation, and rheumatologic markers. Nerve conduction studies (NCSs)/electromyelogram (EMG) was in keeping with generalized severe sensorimotor polyneuropathy and multiple cranial neuropathies. Study of the blink reflex revealed an absent R1 on both sides and prolonged contralateral R2 with stimulation at the right supraorbital notch. Autonomic reflex screen showed postganglionic sudomotor impairment in the lower extremity and mild to moderate cardiovagal impairment. Gastrointestinal esophagram showed penetration of thin liquid barium to the level of the vocal cords. Abdominal fat aspirate revealed amyloid. Transthoracic echocardiogram was performed, and there was no echocardiographic evidence of cardiac amyloid, with normal cardiac wall thickness and left ventricular ejection fraction. A biopsied sural nerve had amyloid deposition predominantly affecting the perineurium and the muscular layer of epineurial blood vessels (Fig. 34-3). Familial amyloidosis DNA sequencing was negative for a mutation in the transthyretin gene. Gelsolin sequencing was performed and the Asp187Asn substitution was identified.
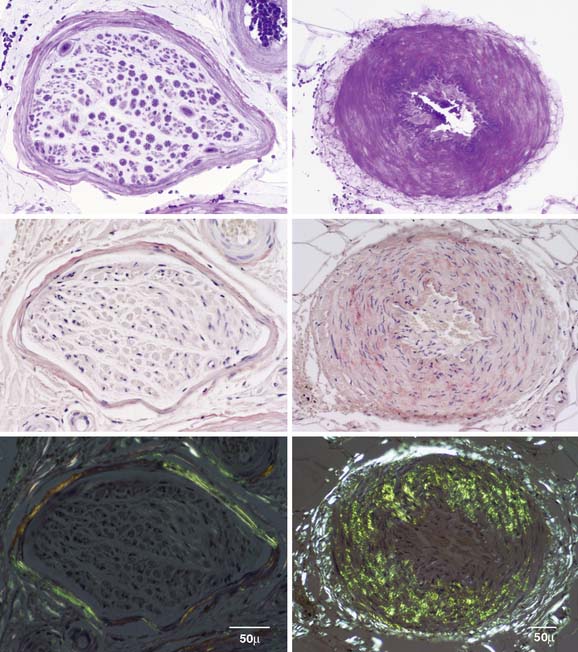
Figure 34-3 Sural nerve paraffin sections. Sections of a nerve fascicle (left column) and epineurial artery (right column) were stained with methyl violet (top row). Middle and bottom rows were stained with alkaline Congo red. Bottom row was viewed under polarized light. Whereas amyloid was visualized under light microscopy in top and middle rows, it is not clearly evident in these figures. By contrast the bottom row shows apple green birefringence typical of amyloidosis.
CONCLUSION
Gelsolin amyloidosis is a rare form of familial amyloidosis, which was first described by Meretoja in 19691 as a type of amyloidosis characterized by cranial neuropathies, corneal dystrophy, and skin changes, among other distinct findings. Gelsolin amyloidosis was originally and is most commonly described within Finnish kindreds but has also been observed in other ethnic groups, including Dutch, Czech, Japanese, and American kindreds. It has an autosomal dominant inheritance pattern, like other forms of familial amyloidosis. The most common type of familial amyloidotic polyneuropathy (FAP) syndrome is transthyretin amyloidosis. Transthyretin amyloidosis has a characteristic pattern of neuropathy with frequent autonomic and small fiber involvement, as well as a length-dependent sensory greater than motor polyneuropathy, and often limits the patient’s life span because of significant cardiac involvement, with arrhythmias and heart failure. Gelsolin amyloidosis, however, tends to present differently, as demonstrated in the above-mentioned patient and in her family. Often the earliest finding in these patients is corneal lattice dystrophy, which appears as lace-like lines of amyloid, primarily along peripheral areas of the cornea, which can be asymptomatic or may result in light sensitivity, corneal erosions, and vision loss.2 Neurologic findings are more limited than in other forms of FAP, with prominent cranial nerve and bulbar dysfunction. The typical neuropathic findings are usually observed by the 4th or 5th decade of life, with slowly progressive facial nerve involvement, and with difficulty raising eyebrows and smiling. This is followed by involvement of other cranial nerves and may result in abnormal facial sensation, decreased hearing, and difficulty swallowing. Laxity of the skin is almost always apparent in these patients, most notably involving the face.2 The progression of disease is slow, and most patients remain alive in their 70s.3
A study of NCS/EMG performed in a cohort of patients with gelsolin amyloidosis showed evidence of cranial nerve dysfunction in all; carpal tunnel syndrome was common, and many of these patients had evidence of peripheral neuropathy with both axonal and demyelinating features.4 Autonomic studies show decreased heart rate variability in gelsolin amyloidosis patients compared with healthy control patients, but it is rare to have clinically significant amyloid involvement of the heart or findings of amyloid infiltration on echocardiography.5 Because of reports of abnormal central nervous system findings in these patients, such as nonverbal abnormalities on neuropsychological testing, punctate leukoencephalopathy on magnetic resonance imaging scan, and abnormal evoked potentials,6 postmortem evaluation of the brains and spinal cords of four patients was performed and extravascular deposits of amyloid were identified in dura, spinal nerve roots, and sensory ganglia,7 which indicate that there may be a more systemic distribution of amyloid than clinical symptoms suggest.
Amyloid can be produced by many different proteins, one of which is gelsolin, which form an abnormal beta-pleated sheet conformation and deposit within otherwise normal tissue. Gelsolin is an actin-modulating protein produced in many different tissues within the body, which can become amyloidogenic when destabilized and cleaved. The mutations identified in gelsolin amyloidosis, D187N and D187Y (asparagine or tyrosine substituted for aspartic acid), lead to decreased calcium binding to domain 2 of the protein, thereby making the structure more susceptible to cleavage. The abnormal protein is then cleaved by the protease furin, in the Golgi complex, into peptides, which deposit together as amyloid in susceptible tissues.8 Treatment is limited, with no clinically available strategies to prevent ongoing amyloid deposition. Symptomatic therapies are employed, such as lubrication of the eyes and reconstructive surgery for facial abnormalities. It has been suggested that either the development of furin inhibitors or “chemical chaperoning” to stabilize the abnormal gelsolin may ultimately be useful strategies for the treatment of this disorder.9
Related posts:
 An Unusual Case of Pia-Arachnoiditis due to Metastatic Breast Carcinoma
An Unusual Case of Pia-Arachnoiditis due to Metastatic Breast Carcinoma
 Stepwise Facial and Upper Limb Weakness and Small Fiber Sensory Loss—Tangier Disease
Stepwise Facial and Upper Limb Weakness and Small Fiber Sensory Loss—Tangier Disease
 Late-Onset Transthyretin Val30Met Familial Amyloid Polyneuropathy Unrelated to Endemic Foci
Late-Onset Transthyretin Val30Met Familial Amyloid Polyneuropathy Unrelated to Endemic Foci
 Inflammatory Multifocal Encephalomyelopolyradiculoneuropathy Mimicking a Granulomatous or Lymphomatous Process
Inflammatory Multifocal Encephalomyelopolyradiculoneuropathy Mimicking a Granulomatous or Lymphomatous Process
 Chronic Inflammatory Demyelinating Polyradiculoneuropathy–Like Neuropathy with Gastrointestinal Dysmotility, Ophthalmoplegia, and Leukoencephalopathy—Mitochondrial Neurogastrointestinal Encephalopathy
Chronic Inflammatory Demyelinating Polyradiculoneuropathy–Like Neuropathy with Gastrointestinal Dysmotility, Ophthalmoplegia, and Leukoencephalopathy—Mitochondrial Neurogastrointestinal Encephalopathy
 “Small-Fiber” Neuropathy, Angiokeratoma, and Heat Intolerance—Fabry’s Disease
“Small-Fiber” Neuropathy, Angiokeratoma, and Heat Intolerance—Fabry’s Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


