CHAPTER 135 Craniopharyngioma
“Craniopharyngioma” was the name introduced by Cushing for tumors derived “from epithelial rests ascribable to an imperfect closure of the hypophysial or craniopharyngeal duct.”1 It remains a rare but challenging tumor given that its location, difficulties associated with treatment, and a high recurrence rate can lead to significant morbidity in patients whom it affects. The treatment and general therapeutic approach to craniopharyngioma have undergone several transformations, even within the modern neurosurgical era, with proponents of both radicalism and conservatism. Significant advances in preoperative imaging, surgical techniques, and adjuvant therapies have enabled neurosurgeons and neuro-oncologists to improve the quality of care that these patients receive.
Historical Review
One of the earliest descriptions of craniopharyngioma is credited to Zenker, who recognized a suprasellar lesion containing cholesterol crystals in an 1857 autopsy study.2 In 1892, Onanoff coined the term pituitary adamantinoma after appreciating the similarities between tumors of the jaw and tumors of the pituitary region.3 In 1899, the pathologists Mott and Barrett began to investigate a group of epithelial-type tumors that occupied the sellar region. They postulated that these tumors arose from either Rathke’s pouch or the hypophysial duct.4 In the next few years, these tumors were reported by both Babinski and Frohlich as suprasellar lesions without acromegaly.5,6 In 1909, Halsteadt was credited with the first transsphenoidal removal of a craniopharyngioma from a patient with symptoms of a sellar mass.7
These tumors were initially named craniopharyngeal pouch tumors in 1921 by McKenzie and Sosman.8 Other terms such as ameloblastoma, epidermoid tumor, and craniopharyngeal fat tumor were also applied before Frazier, Alpers, and Cushing began to use the term craniopharyngioma.1,9 This term is now well entrenched in the neurosurgical literature, although embryologically these tumors are remnants of the primitive stomodeum and not the pharynx.10
The surgical philosophy regarding the treatment of craniopharyngioma has also vacillated significantly over the past 5 decades. Early operative series demonstrated an extremely high mortality rate of 40%, with only 15% of patients undergoing total removal.11 By the early 1960s, many believed that aggressive surgery should be abandoned in favor of cytoreduction combined with radiotherapy.12 In the mid-1970s, with improvements in both postoperative endocrinologic care and overall surgical technique, there was renewed support for an aggressive surgical approach.13–15 Even today, controversy exists between those who advocate aggressive surgical resection and those who support a more conservative approach.
Craniopharyngioma is thought to arise from ectodermally derived epithelial cell remnants of Rathke’s pouch and the craniopharyngeal duct. Neoplastic transformation of cells derived from tooth primordia gives rise to adamantinomatous craniopharyngioma, whereas such transformation in cells derived from buccal mucosa primordia gives rise to the papillary type.16 By the fourth week of gestation, invagination of the stomodeum, lined by epithelial cells, takes place. This upward migration is met by a downward movement of neuroepithelium from the hypothalamus. This upward invagination, termed Rathke’s pouch, is responsible for development of the adenohypophysis, whereas the downward growth of neuroepithelium is the precursor of the future neurohypophysis.
As Rathke’s pouch meets the infundibulum, it separates from the stomodeum and rotates around the anterolateral surface of the infundibulum. This rotation, which occurs during formation of the adenohypophysis, is responsible for delivering embryonic rests to suprasellar or parasellar locations. This migration pathway from the primitive oral cavity is termed the craniopharyngeal duct. In 1904, Erdheim reported that the origin of craniopharyngiomas was based on incomplete involution of this pathway.17
Recent evidence supports the hypothesis that embryonic rests of cells from the craniopharyngeal duct produce the pituitary gland, Rathke’s pouch, and craniopharyngiomas.18 Both human chorionic gonadotropin and P-glycoprotein have been demonstrated to be produced by all these structures.18,19
Challenges to the craniopharyngeal duct hypothesis have centered on evidence that children younger than 10 years rarely have any squamous cell rests around the pituitary. It has been demonstrated that only 3% of neonates harbor these cell rests; however, they are found more often in older individuals.20,21
The second major hypothesis proposed for the pathogenesis of craniopharyngioma centers on the premise that existing cell rests in the adenohypophysis undergo metaplasia. This concept partially accounts for the observation that squamous papillary tumors occur predominantly in adults and lack any resemblance to tooth-forming epithelium. Arguments against this hypothesis are based on evidence that mixed tumors demonstrating both adamantinomatous and papillary squamous characteristics exist.22,23
Incidence
Craniopharyngiomas are relatively uncommon tumors, with the annual incidence rate being between 0.5 and 2.5 new cases per million population per year.24 Craniopharyngioma has a higher annual incidence of 5.25 cases per million in the pediatric population,25 in whom it accounts for 6% to 13% of intracranial tumors. In 60% of patients, craniopharyngioma is diagnosed after the age of 16.26–29
Age
Craniopharyngioma has two histopathologic subtypes, with significant implications regarding age at diagnosis. Adamantinomatous craniopharyngioma is observed to have a bimodal distribution, with one peak in children between 5 and 15 years old and a second peak in adults 45 to 60 years old. The papillary subtype occurs almost exclusively in adults at a mean age of 40 to 55.24,30,31
Sex
Most large series do not show any consistent sex predilection for craniopharyngiomas. Some studies have shown a greater preponderance in males in childhood and in females in adulthood,24,32 and a large series in England suggested that males are affected 30% more often than females.33
Location
Craniopharyngiomas may arise anywhere along the craniopharyngeal duct, but they most commonly arise in the sellar/parasellar region. Four percent are purely intrasellar, 21% are sellar and suprasellar, and 75% are suprasellar alone, often with extension up into the third ventricle.34–36 In very rare cases, craniopharyngiomas may develop in an ectopic fashion, with reports of sites such as the sphenoid sinus,37 nasopharynx,38 clivus,39 and extradural temporal region.40
Geography
Geographic variation may also exist, with various reports citing craniopharyngiomas as accounting for 1.5% to 6.5% of all primary brain tumors. The low incidence of 1.5% in Australia contrasts with the 6.5% rate in China and a possibly higher incidence in Africa.41–43 An American study examining three different databases calculated similar incidence rates of 0.13 per 100,000 person-years.24 Other groups have published comparable incidence rates of 0.5 to 2.0 new cases per year per million population.32,44
Given the predominant location of these tumors in the sellar/parasellar region, craniopharyngioma is characteristically manifested as three clinical syndromes: visual dysfunction, disturbance of the hypothalamic-pituitary axis, and raised intracranial pressure as a result of obstruction of flow of cerebrospinal fluid (CSF) and hydrocephalus.26,45–47 The median duration of symptoms in a series of 34 adult patients was 10 months. The pediatric cohort of patients in this mixed series had a median symptom duration of just 3 months.48 Adults are more likely than children to have symptoms of visual or endocrine abnormalities, whereas children more often have symptoms of increased intracranial pressure. In adults, headache, vomiting, and visual disturbance (hemianopia, uniocular visual loss, and diplopia) are the most common initial complaints.49 More than 80% of adult patients complain of some visual loss at initial evaluation and have evidence of a visual deficit on formal testing. In children, the most common initial symptom is headache, which occurs in 50% to 80%,26,50 followed by vomiting (21% to 68%) and visual deterioration (47% to 80%).
The most common neurological signs relate to visual disturbance: visual field defects (35% to 79%), papilledema (10% to 50%), optic atrophy, and eye movement disorders. In adult patients, 29% have evidence of papilledema at initial encounter, compared with more than 50% in the pediatric population.48 In very young children, increased head circumference or a bulging fontanelle may be seen. Ataxia is described in about 20% of instances.
Approximately 30% of adults will initially have symptoms of endocrine disturbance.51 Gonadal insufficiency is the most common endocrine abnormality at diagnosis and consists of loss of libido and reduced masculine hair growth pattern in men. Women may complain of irregular menstrual periods or even amenorrhea. Other endocrinologic problems at initial evaluation include diabetes insipidus, hyperprolactinemia, adrenal insufficiency, and thyroid insufficiency.
Less than 15% of children with craniopharyngioma have complaints attributable to an endocrinologic deficit,52 even though almost 90% have some endocrine abnormality. Delayed puberty (4% to 24% of patients), obesity (8% to 15%), anorexia, short stature, and precocious puberty are all potential manifestations of craniopharyngioma.23,26,53 Growth hormone deficiency is the most frequently observed deficit, with more than 75% of patients being affected. Specific hormonal deficiencies are identified, with luteinizing hormone/follicle-stimulating hormone (40% of patients) more commonly affected than adrenocorticotropic hormone (25%) and thyroid-stimulating hormone (25%).54 Diabetes insipidus is reported in up to 17% of children and 30% of adults.
Hydrocephalus is an important concomitant factor, particularly in pediatric craniopharyngioma. Hydrocephalus is identified at diagnosis in approximately a third of craniopharyngioma patients overall and in almost one third of children with craniopharyngioma36,54–56 and may require definitive treatment if primary tumor surgery fails to resolve the issue. In one series, 43% of children went on to require long-term treatment of hydrocephalus.56 In another series, only 29% of adult patients had any evidence of hydrocephalus at initial evaluation, although there was a greater than 50% incidence of hydrocephalus in the pediatric population.48 Adequate treatment of hydrocephalus is imperative to minimize long-term cognitive deficits in these children, especially in those undergoing radiation therapy. There would seem to be a correlation between shunt requirement and worse outcome; however, it remains to be clarified whether this is directly related to shunting or is due to the presence of generally larger tumors with hypothalamic involvement in this group.54
Neurobehavioral abnormalities appear to be more common in adults than in children. It has been estimated that more than 30% of adult patients with craniopharyngioma older than 45 years have dementia or suffer from intermittent confusion, hypersomnia, apathy, or depression.57,58 Children may also suffer from neurocognitive decline and exhibit common clinical features such as abulia, psychomotor retardation, and flattening of affect.
Very rarely, craniopharyngioma may develop acutely after intratumoral hemorrhage59 and rarely may rupture and result in aseptic meningitis or spontaneous drainage through the nasopharynx.60–62
There is considerable difficulty in diagnosing craniopharyngioma in young children because some may have relatively nonspecific symptoms such as vomiting and irritability, and visual field loss is often overlooked. A high degree of clinical suspicion is needed in such cases. It has been recognized that the diagnosis of brain tumors in children is frequently delayed in comparison to other childhood tumors.63–65
In 2007, the World Health Organization (WHO) grading system for craniopharyngioma defined two major subtypes of the tumor, adamantinomatous and papillary, with both corresponding to WHO grade I.66 Transitional or mixed forms have also been described but are rare.30,67 The classic adamantinomatous type appears to be present in more than 95% of pediatric cases.55 It is distinctly uncommon for children to display the papillary squamous subtype in isolation or mixed with adamantinomatous histology. The papillary squamous variant is almost exclusively seen in adults and represents close to 30% of all craniopharyngiomas seen in this population.30,55
Histologically, adamantinomatous craniopharyngioma is composed of squamous epithelium in cords, lobules, and trabeculae surrounded by palisaded columnar epithelium.68 “Wet keratin” nodules may be found within the solid portion of the tumor, and a gliotic reaction with abundant Rosenthal fibers is often seen in adjacent brain tissue (Fig. 135-1).
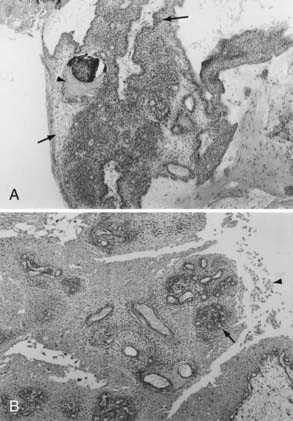
The typical papillary subtype has a well-circumscribed solid appearance with a lower likelihood of cysts and absence of calcification and cholesterol deposits. Histologically, the papillary subtype is a bland mass of well-differentiated squamous epithelium forming pseudopapillae with an anastomosing fibrovascular stroma.30,31,55
Electron microscopy is rarely needed for the diagnosis of craniopharyngioma but typically reveals glycogen and tonofilaments in epithelial cells, which are joined by desmosomes.69 Mineral precipitates are seen in membrane-bound vesicles. Immunohistochemistry is positive for cytokeratins70 and epithelial membrane antigen.68
Malignant transformation of craniopharyngioma has been discussed in the literature but is thought to be very rare and may require additional oncogenic insults, such as exposure to radiation.71–73 The molecular genetic abnormalities associated with craniopharyngioma remain poorly characterized, although there are several reports of clustering of craniopharyngioma in families suggestive of a putative genetic basis.74–76 Some craniopharyngiomas are monoclonal in origin,77,78 and cytogenetic abnormalities have been reported in chromosomes 2 and 12,77,79,80 but additional studies have not shown significant chromosomal imbalances in either type of craniopharyngioma.81,82
Evidence is emerging that implicates specific genes and cell signaling pathways in the development of craniopharyngioma. Mutations of the β-catenin gene have been identified in 70% of adamantinomatous craniopharyngiomas,83,84 whereas none were seen in papillary craniopharyngiomas. β-Catenin is a downstream component of the Wnt intracellular signaling pathway involved in proliferation, morphogenesis, and differentiation,83 and it has been postulated that disruption of this pathway may be important in the pathogenesis of adamantinomatous craniopharyngioma.
Information regarding the natural history of craniopharyngioma is sparse because most patients eventually undergo treatment. In an Oxford (U.K.) series, 13 patients did not initially receive treatment, and 92% of them ultimately required treatment for evidence of disease progression during the study period.36
Plain radiography of the skull is now rarely performed in patients suspected of having a craniopharyngioma; however, radiographs may provide information regarding the size and shape of the sella and may demonstrate lesional calcification. Approximately 65% of adults and 90% of children with craniopharyngiomas will have abnormal findings on skull radiographs. Calcification of the tumor is seen in approximately 40% of adults and 85% of children.85 Calcification is not a unique feature of adamantinomatous lesions inasmuch as it has been demonstrated in histologically confirmed papillary tumors.30,55
Today, MRI is more commonly used to fully characterize a craniopharyngioma. Classically, the solid tumor is isointense to hypointense on T1-weighted images, and T2-weighted images show mixed hypointensity or hyperintensity; reticular enhancement is seen after the injection of gadolinium.86 Edema may be identified in adjacent brain tissue or along the optic tract and is useful in differentiating craniopharyngioma from other tumors in this region.87
In a series of 86 adult patients with craniopharyngiomas at the University of Erlangen, 80% of the tumors were heterogeneous in appearance.51 Most of the tumors were either monocystic with a solid component or predominantly solid with a cystic component. A minority were entirely cystic or entirely solid. All these tumors were suprasellar, with 57% of them harboring an intrasellar component. In addition, 60% of the suprasellar tumors had a component that was retrosellar, and 40% had a component that was in either the posterior fossa or the parasellar space.
There are no radiologic features that can absolutely discriminate among the subtypes of craniopharyngioma. Yet lobulated shape, vessel encasement, and calcification have all been postulated to be indicative of the adamantinomatous subtype.30,67,88
Classification and Grading Schemes
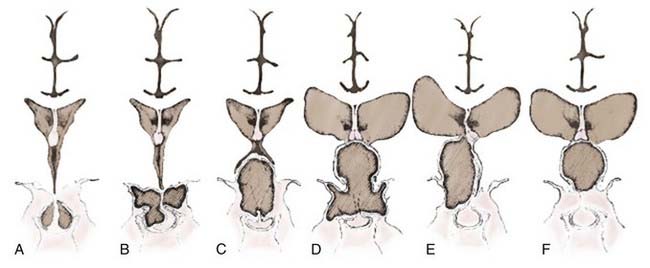
A number of classification schemes have been proposed to assist in decision making regarding surgery, particularly concerning the approach and its concomitant risks.54,89–91 Hoffman and colleagues proposed intrasellar, prechiasmatic, and retrochiasmatic as three basic subtypes,15 whereas Samii and Bini devised a five-tier grading system based on vertical height of the tumor.92 Yasargil and associates recommended grading with respect to the diaphragma and the third ventricle (Fig. 135-2).54 The most recently proposed grading system, from Puget and coworkers, relates to the degree of involvement of the hypothalamus to further define resectability (Fig. 135-3).90 No one system has come into widespread use, but all have useful elements for approaching these tumors surgically, considering their position in relation to the chiasm, or taking into account their vertical height and hypothalamic involvement.
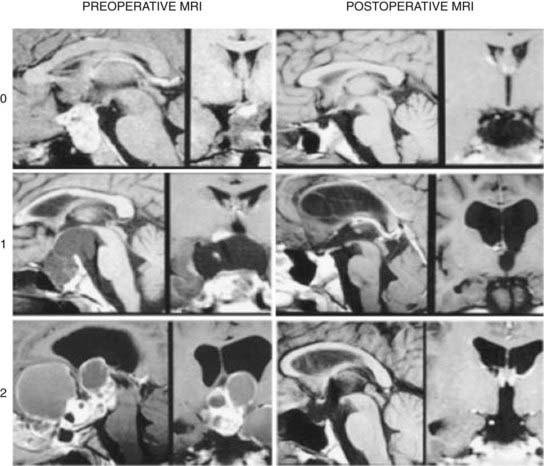
Giant craniopharyngiomas have been defined as tumors that have a maximal diameter greater than 5 cm.93 They are frequently cystic in nature and may extend into the anterior, middle, and posterior fossa.
Differential Diagnosis
Treatment
Full endocrinologic assessment is required, and appropriate replacement therapy should be provided, with particular emphasis on identifying diabetes insipidus and hypocortisolemia. An estimate of bone age and, in young women, ovarian ultrasonography are advocated.94 Likewise, accurate neuro-ophthalmologic assessment, including visual field testing, is critical to have a thorough understanding of the tumor and its relationship to the visual apparatus and as a baseline for analysis after treatment.
Traditionally, surgical resection has been the preferred first option for treatment of these lesions. Most would recommend that if complete surgical resection can be performed without injury to the anterior hypothalamus and without effect on personality and quality of life, it should be performed. The difficulty lies in achieving this goal in a high percentage of cases. An increasing number of experts advocate subtotal resection because of its lower perioperative complication rate, supplemented by adjuvant radiotherapy.56,90,95 Such management has been shown to be associated with equivalent cognitive outcomes and less neurological impairment and deterioration of quality of life.
Occasionally, one may encounter an adult patient who is asymptomatic and found to have a craniopharyngioma on imaging studies. The overall age and medical status of the patient, along with the imaging characteristics of the tumor, are important factors when considering whether treatment is necessary. If one is quite certain about the pathology and the tumor is not threatening the visual apparatus, a period of observation may be warranted. Careful endocrinologic, ophthalmologic, and imaging follow-up can then be used to assess the need for treatment. In a series of 85 predominantly adult patients seen between 1938 and 1970, Bartlett used observation as an approach in 11 patients.57 Five of these patients were monitored for more than 15 years and required no treatment.
Complete surgical resection appears to be an important prognostic factor for recurrence and is the best management method for craniopharyngioma if it can be done safely. It requires great surgical skill and judgment, but considerable effort should be exerted to perform as complete a resection as safely possible. Unfortunately, complete resection may be very difficult to achieve.96
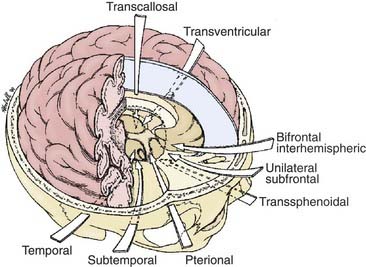
The most appropriate surgical corridor for resecting a craniopharyngioma is determined by numerous factors, including the shape and size of tumor, the presence of cysts, its location, the degree of calcification, and the surgeon’s preference and experience (Fig. 135-4). The standard approaches for attempted GTR include pterional, orbitozygomatic, subfrontal/trans–lamina terminalis, interhemispheric-transcallosal, and transcortical-transventricular; reports of endoscopic endonasal transsphenoidal removal of craniopharyngiomas are emerging as well.
Gross-Total Resection
Subfrontal/Trans–Lamina Terminalis Approach
Suitability
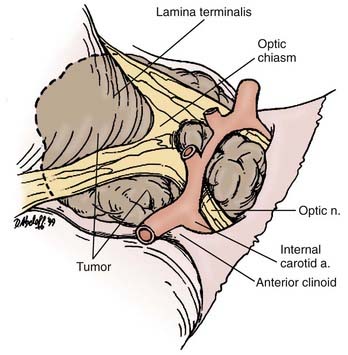
This approach is most favored in the presence of a midline prechiasmatic tumor that extends into the anterior cranial fossa floor or superiorly into the suprasellar cistern and the third ventricle.97 Its advantages include the midline orientation of the approach with access to both the optic nerves and internal carotid arteries, as well as the third ventricle via the lamina terminalis.
Operative Details
The patient is placed in a supine position with the head slightly extended such that the anterior skull base is perpendicular to the floor. Head fixation is preferred, and the use of neuronavigation equipment may be helpful, particularly for solid tumors. A lumbar drain may be considered. Mannitol and dexamethasone are administered. A bicoronal incision is made, with preservation of a vascularized pericranial graft for use in reconstruction if necessary. Once adequate bony exposure has been achieved, a low unilateral or bilateral frontal craniotomy is performed, and if present (in older children and adults), the frontal sinus is opened. In general, we prefer a unilateral craniotomy to avoid bihemispheric injury. Cranialization may need to be performed on the frontal sinus. Additional removal of the orbital bar may provide greater exposure.98 The dural opening is created behind the orbital bar, and the right olfactory nerve will usually be dissected from the undersurface of the frontal lobe or divided. Elevation of the frontal lobes from the anterior cranial fossa floor may be added by dividing the superior sagittal sinus at the floor. Exposure of the optic nerves, optic chiasm, and carotid arteries bilaterally is then afforded. Opening of the lamina terminalis immediately posterior to the chiasm then allows access to tumors that extend into the third ventricle.
Difficulties
Both frontal lobes are elevated during the bifrontal approach, which may be associated with increased postoperative morbidity. This approach is also compromised by the potential for injury or the necessity for sacrifice of one of the olfactory nerves. The frontal sinus will be opened when present (in older children and adults) and will require cranialization; there is a risk for infection or CSF leakage with this procedure. Removal of an intrasellar tumor can be difficult when using a subfrontal approach, but it is possible and may be facilitated by the use of an angled endoscope (Fig. 135-5).
Bifrontal Basal Interhemispheric Approach
Suitability
Although the subfrontal approach is favored by many neurosurgeons, the basal bifrontal interhemispheric approach has been proposed by others.99 This approach is well suited for large, midline, retrochiasmatic tumors that may have retrosellar extension. Although it may be more technically challenging, this approach allows wider visualization of the optic pathway and anterior circle of Willis. Proponents of the bifrontal basal interhemispheric approach cite its ability to avoid the potential blind spots encountered with unilateral approaches. This technique has also been combined with division of the anterior communicating artery in an effort to maximize removal of the retrochiasmatic portion of these tumors.99
Transsphenoidal and Extended Transsphenoidal Approaches
Suitability
Smaller midline tumors within the sella or with an infradiaphragmatic suprasellar component may be removed by the transsphenoidal route. This route is most favorable for patients with enlargement of the sella. Significant suprasellar extension of craniopharyngiomas can be difficult to access from a transsphenoidal approach; however, the presence of cystic rather than solid disease makes its use more favorable.100 Advantages of this approach include the lack of need for brain retraction and potentially better visual outcomes than observed when using cranial approaches.54,101,102
Operative Details
The patient is placed supine on the operating table with the head elevated and slightly angled toward the surgeon. Use of a Mayfield headrest and neuronavigation devices can obviate the need for fluoroscopy, although either or both can be used. The procedure can be performed with an endonasal endoscope103 or by using the operating microscope with a nasal speculum. Sublabial, transseptal, and direct approaches to the sphenoid face have all been described. The mucosa is reflected, and the sphenoid sinus is opened with a drill or Kerrison punch. The mucosa of the sinus is excised, and the anterior wall of the sella is removed to expose the sella dura. Anterior compression of the anterior pituitary is a common finding, and the gland may need to be divided to provide access to the tumor.104
For the extended transsphenoidal procedure, the tuberculum sellae can be removed, and additionally, excision of the planum sphenoidale will provide improved access to the suprasellar region. The dura may then be opened over the gland and the circular sinus and anterior to the sella to provide access to the suprasellar cistern. The dural defect is closed with a graft and supported by bone or synthetic plates, with fat placed in the sphenoid and a vascularized nasal flap added for coverage of the site of surgical access from the nasopharynx.105 Some authors advocate the use of postoperative lumbar drainage to reduce the risk for delayed CSF rhinorrhea.106
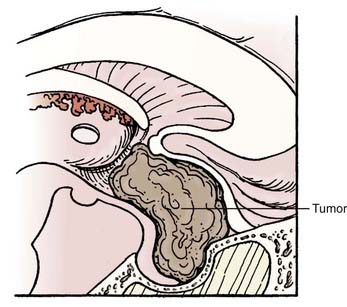
Difficulties
Lateral extension of the tumor can be difficult to access from this approach, as can large tumors. Tumor involvement in the region of the anterior cerebral complex may expose the perforating vessels to risk for injury during surgery. Direct control of intracranial neural and vascular structures is inferior when using the transsphenoidal or extended transsphenoidal approach (Fig. 135-6). Suprasellar calcifications are thought to be a contraindication to the use of this approach.104 This approach can be difficult or impossible to use in children with a poorly pneumatized sinus and in patients with nasal and sinus pathology. Reconstruction after tumor removal can be complicated, and high rates of CSF leakage have been reported in the literature, a problem that has not been completely resolved after extended transsphenoidal surgery. Use of a vascularized nasal mucosal flap is showing promise in this regard.105
Pterional Approach
Suitability
The pterional approach has traditionally been the most favored one for resection of craniopharyngiomas,54,90,96 and it would seem to best be suited for smaller tumors confined to the suprasellar space.98 It does allow access to both prechiasmatic and retrochiasmatic lesions, along with those above and below the diaphragma, and may be used in combination with interhemispheric-transcallosal or transcortical-transventricular approaches to remove larger lesions with significant suprasellar extension.
Interhemispheric-Transcallosal Approach
Suitability
Large midline tumors with suprasellar extension into the third and, potentially, the lateral ventricle can be removed if the interhemispheric-transcallosal approach is combined with a basal procedure. It may be satisfactorily used alone in the rare setting (3% to 10%) of a purely third ventricular craniopharygioma.54






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


