CHAPTER 182 Craniosynostosis
Although skull shape irregularity has been recognized since antiquity, the study of abnormal skull growth related to craniosynostosis had its scientific origin in the late 1700s. Sommerring1 noted that bone growth in the skull occurred primarily at suture lines and that when this growth site was prematurely bridged with bone, an abnormal skull shape developed. This was characterized by reconstruction of the skull’s growth in a plane perpendicular to the plane of the fused suture. Similar observations were made by Otto2 in 1830 and Virchow3 in 1821. It was Virchow’s widely publicized treatise on skull deformity that added impetus to the scientific study of abnormal skull form in craniosynostosis. He observed, as had earlier investigators, that skull growth occurred at suture lines in the skull and that when these suture lines were prematurely fused, skull deformity developed. Restriction of growth adjacent to the suture occurred, but compensatory growth occurred elsewhere in the skull to accommodate the growing brain. This understanding of normal and abnormal skull growth served as the basis for understanding skull abnormalities for the next 100 years (Fig. 182-1).
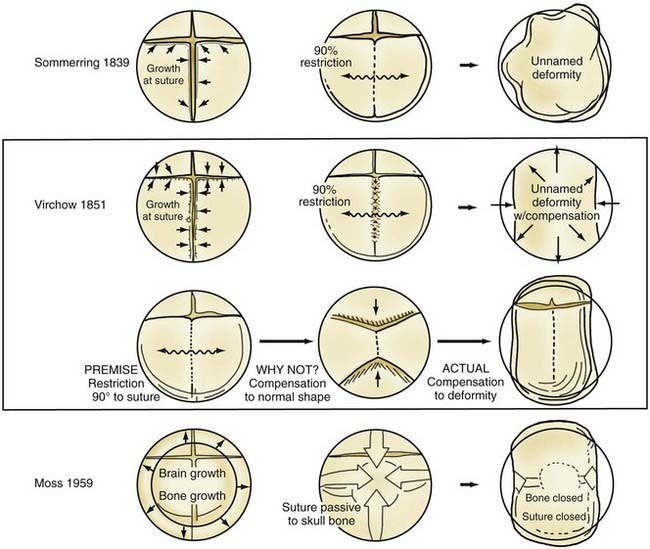
In the mid-20th century, the primary role of the cranial vault suture in the development of craniosynostosis skull deformities was questioned by Moss4 and van der Klaauw.5 Moss noted that surgeons operating on the skulls of children presumed to have craniosynostosis would occasionally find patent cranial vault sutures, despite what appeared to be typical craniosynostosis skull deformities (see Fig. 182-1).6 Also, he recognized that there were some characteristic cranial base deformities associated with individual forms of craniosynostosis, and because the cranial base matures embryologically before the remainder of the cranial vault, he suggested that the cranial base abnormality was the primary pathologic process and the cranial vault suture abnormality (fusion) was secondary.4 Testing this hypothesis in the laboratory, Moss7 removed a normal cranial vault suture and found that doing so did not affect overall skull length. This indicated that the cranial vault suture, unlike the epiphysis of a long bone, does not serve as a growth site that pushes the bone ends apart but is a passive recipient of growth influences.
The amount of bone deposited at the cranial vault suture is related to the strains that influence it. Brain enlargement, Moss8 believed, was the primary source of these tensile strains that caused the suture to deposit bone. This is known as the functional matrix theory, in which the functional enlargement or development of an organ system is the primary force in changing its overall shape and determining its final form. Even though brain enlargement is clearly the engine of skull remodeling, the precise role the vault suture plays in the development of the skull pathology associated with craniosynostosis must be determined by direct manipulation of growth at the suture.7
In 1979 Persson and colleagues9 reported that in animals, selective restriction of an individual cranial vault suture’s growth resulted in skull deformities that closely mimic the clinical condition of craniosynostosis involving the same cranial vault suture in humans. Moreover, cranial base and even facial deformities develop secondary to the cranial vault suture restrictions.10,11 This observation indicates that cranial vault suture pathology may be primary in the development of craniosynostosis skull deformities leading to cranial base and facial deformity. Subsequently, Mooney and coworkers12,13 studied an animal model of congenital craniosynostosis in which cranial vault suture, vault, and cranial base abnormalities closely resembled the findings of Babler and Persing10 on cranial suture restriction. In addition, the developmental studies of Opperman and colleagues14,15 demonstrated the significant influence of mesenchymal tissues, in particular the dura and the periosteum at the suture, on the maintenance of patency of cranial vault sutures during development.
Nonsyndromic and Syndromic Craniosynostosis
Researchers have found clinical evidence that cranial vault suture, not cranial base, pathology is of primary importance in the development of skull shape pathology in nonsyndromic craniosynostosis. Preoperative and postoperative computed tomography (CT) shows that surgical treatment of the cranial vault alone results in improvement of not only cranial vault but also cranial base pathology.16 This indicates that removing the abnormal influence in the cranial vault suture may improve cranial base shape. Further analysis of skull growth after simple suture closure has revealed a predictable pattern based on certain rules. The ability to predict and understand the stereotypical deformity from observation of the vault suture suggests its primary role.17,18 The dura clearly plays a role in this, as demonstrated by Drake and coworkers,19 who found that resected fused and nonfused cranial suture elements redevelop following the removal of cranial bone topographically connected to the fused and nonfused sutures. Distraction devices, such as those developed by Ilizarov for the distraction of extremity bones and by McCarthy for mandibular hypoplasia, are effective in elongating bone in animals. Using spring-like expanders, Persing and associates20 internalized the distraction of skull bone experimentally, and subsequently Maltese and colleagues21 employed this approach in the management of human craniosynostosis. The advantage of the distraction and spring techniques is that bone elongation is a gradual process, with minimal surgical intervention and operative time. One disadvantage is that if the cranial bone is already deformed, the irregularly shaped bone is advanced or expanded from the osteotomy site. In addition, the removal of spring and bone distraction devices requires an operative procedure. Similar to implanted metal fixation plates and screws in the skull, if left in place too long, these metal devices can migrate intracranially, with progressive resorption of the endocranial surface of the skull bone and positioning of bone on the ectocranial surface as the skull and brain enlarge. Concern about transcranial migration has been the main impetus for the avoidance of metallic fixation plates and screws in young children. Resorbable plates and screws, as well as suture material, have largely supplanted titanium devices in patients younger than 3 years.
Additional studies are being done to document skull growth changes by surgical manipulation, in particular by the use of mechanical devices to control or enhance growth influences.17,20 In the future, these devices may be used clinically to prolong corrective growth influences postoperatively.
Syndromic craniosynostosis is much less common and appears to be a more generalized disorder of mesenchymal development that may represent abnormalities in homeobox genes such as Runx2 or CBFA-δ. Crouzon’s syndrome occurs in 1 in 25,000 live births, and this bilateral coronal synostosis is frequently associated with exorbitism and midface hypoplasia. Apert’s syndrome occurs in 1 in 100,000 live births; the features of brachycephaly due to coronal synostosis and midface hypoplasia are evident, but extremity syndactylies are also characteristic. There are more than 64 known craniofacial syndromes associated with craniosynostosis22; however, the cause of craniosynostosis is still unknown, although in some cases, a genetic influence is undeniable. In the syndromic forms of craniosynostosis, autosomal dominant inheritance patterns are the general rule for the more common syndromes such as Apert’s, Crouzon’s, and Pfeiffer’s; autosomal recessive disorders (e.g., Carpenter’s syndrome, characterized by abnormal skull shape deformities and polydactyly) do not commonly occur.
Diagnosis
Nonsyndromic craniosynostosis involving a single vault suture is ordinarily diagnosed by the observation of a typically deformed skull shape, with radiographs serving as confirmatory evidence. We have modified the Virchow hypothesis of skull deformity to explain the skull shapes associated with individual forms of craniosynostosis.17,18 These patterns of skull abnormalities can be explained by invoking four tenets: (1) cranial vault bones directly adjacent to the prematurely fused vault suture act as a single bone plate, with reduced growth potential along all margins of that plate (Fig. 182-2A); (2) asymmetric bone deposition occurs at vault sutures along the perimeter of the bone plate, with increased bone deposition occurring at the suture margin located farther away from the plate (Fig. 182-2B); (3) the nonperimeter sutures in line with the fused suture deposit bone symmetrically at their sutural edges (Fig. 182-2C); and (4) perimeter and sutures (in line) abutting the prematurely fused suture compensate to a greater degree than distant sutures (Fig. 182-2D). The application of these principles to particular types of synostosis is as follows (Fig. 182-2E to H)  : Figure 182-2E represents sagittal synostosis, in which the coronal and lambdoid sutures show asymmetric growth, and the metopic suture shows symmetrical growth. Figure 182-2F depicts coronal synostosis, with asymmetric growth at the metopic, lambdoid, and sagittal sutures and symmetrical growth at the unfused coronal suture. Figure 182-2G illustrates metopic synostosis, with symmetrical growth at the sagittal suture and asymmetric growth at the coronal sutures. Figure 182-2H depicts lambdoid synostosis, with a fused lambdoid suture and bilateral growth at the lambdoid, unilateral at the sagittal, and unilateral at the squamosal sutures, displacing the ear downward rather than forward as in positional posterior plagiocephaly.
: Figure 182-2E represents sagittal synostosis, in which the coronal and lambdoid sutures show asymmetric growth, and the metopic suture shows symmetrical growth. Figure 182-2F depicts coronal synostosis, with asymmetric growth at the metopic, lambdoid, and sagittal sutures and symmetrical growth at the unfused coronal suture. Figure 182-2G illustrates metopic synostosis, with symmetrical growth at the sagittal suture and asymmetric growth at the coronal sutures. Figure 182-2H depicts lambdoid synostosis, with a fused lambdoid suture and bilateral growth at the lambdoid, unilateral at the sagittal, and unilateral at the squamosal sutures, displacing the ear downward rather than forward as in positional posterior plagiocephaly.
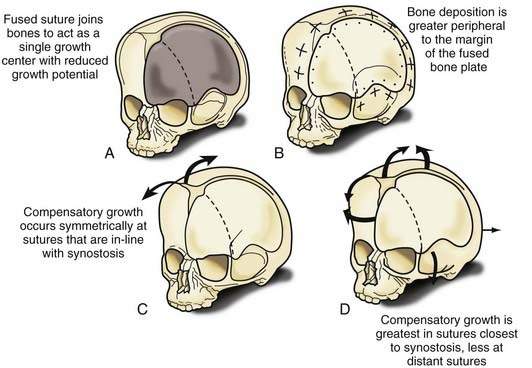
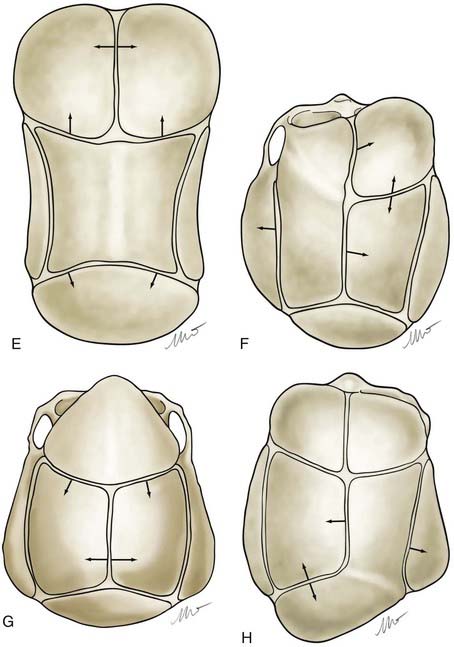
Figure 182-E2 contd E, Sagittal synostosis. F, Coronal synostosis. G, Metopic synostosis. H, Lambdoid synostosis.
(E-H, Courtesy of Dr. Michael McKisic.)
There is controversy regarding the advisability of CT scans in patients being evaluated for craniosynostosis. It has been reported that CT scans performed early in infancy are associated with an increased likelihood of malignancy later in life,23 although the concern for malignancy is greatest in children undergoing thoracoabdominal scans and in those scanned earlier in life.
Operative Timing and Approaches
A common question asked by parents is whether the condition is progressive—in other words, will it “look worse” with time? This is particularly pertinent in sagittal synostosis, where the answer appears to be affirmative. Serial CT imaging has shown that normal growth of the skull tends toward increasing roundness in the first year of life. Sagittal synostosis results in a progressively longer skull, but because the width also increases, the shape does not change as it should, resulting in a marked increase in the deformity.24
Experimental evidence demonstrates that improved skull form is achieved with an operation before the human equivalent of 6 months of age.25 In clinical studies, more significant degrees of improvement were noted in patients who underwent surgery when younger than 6 months compared with those having surgery after 6 months of age. Correction of the cranial base deformities established in unilateral coronal synostosis was also more successful if surgery was performed early.16
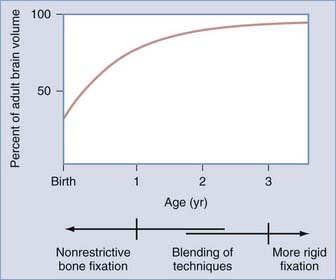
The influence of surgical methods is not yet clearly defined; however, limited craniectomy procedures of few sutures appear to be beneficial in very young patients with mild deformities, whereas more comprehensive approaches to established skull deformities are more effective in older children and in children of any age with more severe deformities. Objective documentation of these approaches is not yet widespread, but Marsh and coworkers25 compared preoperative and postoperative skull shape following a limited craniectomy procedure versus a whole-vault cranioplasty approach for sagittal synostosis; they found that skull deformity was reduced with both craniectomy and cranioplasty procedures. Heller and associates26 showed a normal cephalic index in 100% of patients undergoing whole-vault cranioplasty; however, the skull shape returned to normal only in patients who underwent the more extensive cranioplasty procedure. Additional data are required to confirm initial impressions, but they are consistent with the hypothesis that early rapid growth of the brain shapes the skull (Fig. 182-3).
Intracranial Pressure, Cognition, and Behavior
Treatment of brain deficiency by surgical attack on the deformed skull has an inglorious history dating to the second half of the 19th century. Many luminaries of the early years of neurosurgery were caught up in an international wave of unscientific enthusiasm for linear craniectomy as a therapy to “unlock” the brains of severely impaired, microcephalic children. The benefits were imaginary, and the mortality rates were appalling. A passionate campaign led by Abraham Jacobi, patriarch of the specialty of pediatrics and founder of the American Academy of Pediatrics, and supported by Harvey Cushing, among others, eventually drove the surgical treatment of mental retardation into deserved obscurity (see Feinsod and Davis27 for the full story). When craniosynostosis later emerged as a distinct diagnostic entity, and as surgical interventions became safe and effective from a cosmetic standpoint, the question of the relationship between skull deformity and brain development arose once again, and it has proved resistant to simple answers.
Elevated intracranial pressure (ICP) is a well-recognized feature of the syndromic forms of craniosynostosis, but it can be documented in a fraction of cases of single-suture synostosis as well.28–40 Generally it subsides after surgical treatment.36,39,41 ICP monitoring has been proposed as an adjunct to surgical decision-making in cases for which cosmetic indications are not compelling.49 The most extensive experience with ICP monitoring comes from the Hopital des Enfants Malades in Paris.30,32,38,40 In a 1989 report of 358 ICP recordings in children with craniosynostosis, Renier32 took ICP recorded during slow-wave sleep as “baseline.” In the absence of valid age-adjusted, normative ICP data, baseline measurements less than 10 torr were defined as normal. Measurements above 15 torr were considered abnormal, and measurements between 10 and 15 torr were considered borderline. Elevated ICP was recorded in 8% of cases of sagittal synostosis, 6% of metopic synostosis, and 12% of unilateral coronal synostosis. It was more prevalent in patients with multiple suture involvement and among syndromic cases. Patients who presented after 1 year of age had much higher rates of intracranial hypertension. Likewise, patients who presented after 1 year of age had lower developmental quotients (DQs) or intelligence quotients (IQs), and there was an inverse correlation between preoperative ICP and DQ or IQ.
The generalization of these data in support of early surgical treatment is difficult to accept. Patients were selected for ICP monitoring for unstated reasons, and the factors determining early and late presentation were unknown as well. It is possible that the parents of the older patients found their children’s deformities acceptable but brought them to medical attention because of developmental or behavioral issues—a selection bias that may have enriched the late presenting group with low DQs and IQs.40 Another possibility is that late presenting cases had experienced obstacles to access to care based on low socioeconomic status or low parental intelligence. This analysis has also been criticized for the interchangeable use of DQ and IQ data, because longitudinal studies have shown that DQ scores above the range of frank retardation are only weakly predictive of IQ later in life.40,42 The association of single-suture craniosynostosis with intracranial hypertension is further clouded by mechanistic questions. Perhaps contrary to expectations, measurements of cranial volume using CT have documented normal or increased volumes among infants with single-suture craniosynostosis compared with age-matched controls.43–45 The correlation between intracranial hypertension and lower than normal cranial volume seems to be quite poor, and the measurement of normal or even increased cranial volume does not eliminate the possibility of elevated ICP.29,30 As is true among patients with syndromic craniosynostosis, mechanisms other than craniocerebral disproportion, such as cerebral venous insufficiency, may be important.
A few studies have reported focal regions of hypoperfusion46,47 or hypometabolism48 subjacent to abnormal sutures among small numbers of selected infants with single-suture craniosynostosis. In most cases these abnormalities resolved after surgery. The prevalence of such focal physiologic disturbances cannot be estimated. The natural history and functional significance of such disturbances are unknown.
Recent investigations employing sophisticated methods of image analysis have begun to attempt to relate calvarial deformities to deformation of the underlying neuroanatomic structures and to development data.49–53 Efforts to demonstrate correlations between preoperative skull morphology and preoperative development testing have yielded contradictory findings.51–54 Distinctive alterations in cortical and subcortical brain morphology in association with sagittal and metopic synostosis have been demonstrated on magnetic resonance imaging in comparison with age-matched controls.49 Preoperative and postoperative studies of infants with sagittal synostosis indicate that these distortions of brain morphology do not revert toward normal even after successful surgical correction of the calvarial deformity.50
Since the beginning of the modern era of surgical treatment of craniosynostosis, there have been concerns about the intellectual and behavioral prognosis of affected children,55,56 leading to many detailed observational studies. Kapp-Simon and others have reviewed this literature lucidly.57–59 Points of observation fall into three time periods: at presentation in infancy, early in the postoperative period, and at school age or later. Observations in the first two periods are necessarily developmental. In the last period, intelligence can be tested, and behavioral disturbances and learning disabilities can be recognized. The standard instruments, typically the Bayley Scales of Infant Development and the Wechsler Intelligence Scales for Children—Revised, have age- and gender-adjusted norms, but some studies have employed matched community controls or sibling controls, with particular implications for the interpretation of results. In all three periods, the rates of true mental retardation (DQ or IQ more than 2 standard deviations below the mean) are probably no greater among children with single-suture synostosis than in the general population. Most investigators have found that in study groups of infants with single-suture involvement, the distributions of preoperative developmental test scores are normal or shifted downward to variable degrees, with variable statistical significance in comparison to normative data.52,59–66 In the early postoperative period, aggregate test scores are generally stable,61,62,66,67 although some studies have claimed improvements over preoperative scores that are not enjoyed by untreated patients.60 Later in childhood the differences between patients and unaffected children become more vivid: among patients, mean full-scale IQ scores are within the normal range but generally lower than among controls,68 and a growing fraction of patients are diagnosed with speech, language, or learning disabilities and behavioral disturbances. The literature suggests that between 35% and 50% of children with single-suture synostosis, regardless of treatment status, can be expected to exhibit some degree of cognitive or behavioral disability in the school years.54,59,69–74
Clinical research will never directly address the question of the effect of surgical treatment on the long-term cognitive and behavioral prognosis for children with single-suture craniosynostosis. A randomized trial cannot be conducted. Although there may be equipoise on the question of cognitive and behavioral outcomes, the contemporary standard of care dictates the treatment of almost all patients for cosmetic indications. Observational studies have drawn conflicting inferential conclusions based on the presence or absence of statistical correlations between treatment status or age at treatment and various outcomes of interest.32,40,54,65,72 A host of factors, known and unknown, determine whether and when a child gets surgical treatment, and many of these factors are potential determinants of cognitive and behavioral outcomes as well. Such sources of bias prohibit the confident interpretation of the existing literature.
Operative Treatment
Because the objective of surgical treatment is to provide each patient with the best aesthetic outcome that can be achieved, the plan for correcting the deformity must be formulated according to the particular demands of each case. The technique of surgical reconstruction must be tailored to the type of synostosis present and the age of the patient. It is clear that reshaping bone in a child younger than 1 year is much more readily achieved without significant bone disruption than in an older child. The methods of fixation for reshaped skull segments should also be different for very young children to avoid abnormalities in brain development resulting from vault surgery and the subsequent restriction of brain growth. Patients who are younger than 3 years have a substantial amount of brain growth remaining, but because those older than 3 years have already achieved approximately 85% to 90% of their ultimate brain mass (see Fig. 182-3), the fixation techniques can be more rigid and inclusive in these older children. In the following sections, the treatment of patients younger than 1 year and older than 3 years is described separately; those between 1 and 3 years of age require a blending of techniques. Metallic fixation devices are generally avoided in patients younger than 3 years owing to concerns related to the inward “migration” of the metal with further skull development. Resorbable plates, screws, and sutures are used instead of metallic devices.
Metopic Synostosis
Metopic synostosis is characterized by metopic suture ridging, bilateral flattening of the frontal bones, anterior displacement of the coronal sutures, lateral flaring of the posterior parietal regions, hypotelorism, and flattening of the supraorbital ridges (Fig. 182-4). Viewed from above, the skull has a characteristic triangular shape known as trigonocephaly.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


