Primary dystonia
Typically early-onset: generalized
DYT1 generalized dystonia
Non-DYT1 generalized dystonia
Paroxysmal dystonia and dyskinesias
Dopa-responsive dystonia
Myoclonic dystonia
Rapid-onset dystonia-parkinsonism
X-linked dystonia-parkinsonism
Typically adult-onset: focal
Blephalospasm
Cervical dystonia
Writer’s cramp
Embouchure dystonia
Oromandibular dystonia
Laryngeal dystonia
Secondary dystonia
Medications, trauma, toxins, infections, or stroke.
Pelizaeus-Merzbacher disease
Perinatal cerebral injury
Dystonia-deafness syndrome
Viral encephalitis
MERRF
SSPE
MELAS Leber’s disease
AIDS
Leigh’s syndrome
Creutzfeldt-Jakob disease
Neuroacanthocytosis
Kernicterus
Intranuclear inclusion disease
Huntington’s disease
Hemochromatosis
Parkinson’s disease
Progressive supranuclear palsy
Spinocerebellar ataxias
Multiple system atrophy
HARP syndrome
Corticobasal degeneration
Familial frontotemporal dementias
Dentatorubropalidoluysian atrophy
Familial basal ganglia calcifications
Glutaric academia
Wilson’s disease
Methylmalonic academia
Juvenile parkinsonism
Homocystinuria
Ataxia-telangiectasia
Metachromatic leukodystrophy
Triosephosphate isomerase def
Neuronal ceroid lipofuscinosis
Vitamin E deficiency
Primary antiphospholipid antibody
Biopterin deficiency
Gangliosidoses
Sphingolipidoses
Hallervorden-Spatz disease
Niemann-Pick disease
Multiple sclerosis
Ceroid lipofuscinosis
Atlantoaxial sublaxation
Homocystinuria
Syringomyelia
Hartnup disease
Arnold-Chiari malformation
Methylmalonic aciduria
Congenital Klippel-Feil syndrome
Tyrosinemia
Lesch-Nyhan syndrome
Rett’s syndrome
Table 10.2
Classification of dystonia by age and distribution
Classification by age of onset |
Early onset (childhood and young adulthood, generally <26 years old) |
Late onset (generally ≥26 years old) |
Classification by anatomic distribution |
Focal (involving a single body area) |
Segmental (involving two or more contiguous body areas) |
Generalized (involving at least one leg, the trunk, and another body area) |
Multifocal (involving two or more noncontiguous body areas) |
Hemidystonia (involving one side of the body) |
Table 10.3
List of hereditary dystonias named DYT
Symbol | Gene | Locus | Alternative name |
|---|---|---|---|
DYT1 | TOR1A | 9q34 | Early-onset torsion dystonia |
DYT2 | Unknown | Unknown | Autosomal recessive torsion dystonia |
DYT3 | TAF1 | Xq13 | X-linked dystonia-parkinsonism |
DYT4 | TUBB4 | 19p13.12-13 | Autosomal dominant whispering dysphonia |
DYT5a | GCH1 | 14q22.1-q22.2 | Autosomal dominant dopamine-responsive dystonia |
DYT5b | TH | 11p15.5 | Autosomal recessive dopamine-responsive dystonia |
DYT6 | THAP1 | 8p11.21 | Autosomal dominant dystonia with cranio-cervical predilection |
DYT7 | Unknown | 18p (questionable) | Autosomal dominant primary focal cervical dystonia |
DYT8 | MR1 | 2q35 | Paroxysmal nonkinesigenic dyskinesia |
DYT9 | SLC2A1 | 1p35-p31.3 | Episodic choreoathetosis/spasticity (now known to be synonymous with DYT18) |
DYT10 | PRRT2 | 16p11.2-q12.1 | Paroxysmal kinesigenic dyskinesia |
DYT11 | SGCE | 7q21 | Myoclonic dystonia |
DYT12 | ATP1A3 | 19q12-q13.2 | Rapid onset dystonia parkinsonism and alternating hemiplegia of childhood |
DYT13 | Unknown, near D1S2667 | 1p36.32-p36.13 | Autosomal dominant cranio-cervical/upper limb dystonia in one Italian family |
DYT14 | 14q | Dopa-responsive dystonia | |
DYT15 | Unknown | 18p11 | Myoclonic dystonia not linked to SGCE mutations |
DYT16 | PRKRA | 2q31.3 | Autosomal recessive young onset dystonia parkinsonism |
DYT17 | Unknown, near D20S107 | 20p11.2-q13.12 | Autosomal recessive dystonia in one family |
DYT18 | SLC2A1 | 1p35-p31.3 | Paroxysmal exercise-induced dyskinesia |
DYT19 | Probably PRRT2 | 16q13-q22.1 | Episodic kinesigenic dyskinesia, probably synonymous with DYT10 |
DYT20 | Unknown | 2q31 | Paroxysmal nonkinesigenic dyskinesia |
DYT21 | Unknown | 2q14.3-q21.3 | Late-onset torsion dystonia |
DYT23 | ANO3 | 11p14.2 | Autosomal dominant cranio-cervical dystonia with prominent tremor |
The diagnosis of dystonia is based mainly upon clinical features, although gene analysis can provide supportive evidence. Observation and examination of the patient with detailed clinical history are most important in diagnosis of dystonia. Patients with typical DYT-1 dystonia have normal milestones of development during early childhood, and then symptom starts from a leg or an arm. In the early stage of dystonia, sensory trick phenomenon (simple touching of a certain part of the body improves the symptoms) and morning benefit (symptoms are better immediately after waking up in the morning) are observed. The symptoms are always the same and stereotype. If these three manifestations are confirmed, diagnosis is not difficult. For further details, please refer to the UpToDate (Comella 2013).
10.3 Surgical Indication
It is well established that young-onset DYT-1-positive generalized dystonia is the best indication of pallidal DBS (Panov et al. 2013; Haridas et al. 2011; Isaias et al. 2009), while other non-hereditary adult-onset primary generalized dystonia respond to pallidal DBS as well. It has been shown that younger onset and shorter duration of the disease are the favorable factors of DBS treatment (Yamada et al. 2013; Lumsden et al. 2013). Symptoms of the trunk and proximal parts of extremities seem to respond better than the neck or the distal part of the extremities. This means that patients may have persistent difficulty of writing due to focal hand dystonia even after well controlled other symptoms. Tonic neck dystonia may be a residual symptom after DBS, and peripheral surgical procedure or botulinum toxin injections may be necessary. Speech difficulty may be refractory to DBS.
Patients with phasic or complex type cervical dystonia or orofacial dystonia (Meige syndrome) can be good candidates of pallidal DBS, if the symptoms are refractory to botulinum toxin injections (Kim et al. 2012; Ostrem et al. 2011; Morgan and Sethi 2008; Krauss 2007; Lyons et al. 2010; Witt et al. 2013; Walsh et al. 2013). It is often said that secondary dystonia does not respond well to pallidal DBS as compared with primary dystonia. Although this may be generally true, tardive dystonia due to anti-psychotic medications can be as well controlled as primary dystonias (Eltahawy et al. 2004; Franzini et al. 2005; Cohen et al. 2007; Sako et al. 2008; Gruber et al. 2009; Chang et al. 2010; Trinh et al. 2014). Functional or social outcome of tardive dystonia after DBS depends on the psychiatric state, and therefore medications necessary for their psychiatric symptoms should not be stopped and close follow-up by psychiatrists is mandatory (Mentzel et al. 2012; Jahanshahi et al. 2011). There are few reports on DBS for dystonias of cerebral palsy origin (Katsakiori et al. 2009; Koy et al. 2013; Marks et al. 2013). Because of heterogeneity of the symptoms of cerebral palsy such as mixture of dystonia, spasticity, and athetosis, the effect of pallidal DBS in CP seems modest. However, even with such modest improvement of dystonia, patients may often feel great improvement probably because lifelong motor impairment had been so severe. A few patients with mild CP may develop severe generalized dystonia, and such patients can be good candidates of pallidal DBS.
Many congenital metabolic disorders can induce dystonias. Among them, dystonia in Lesch-Nyhan syndrome (Taira and Hori 2003; Deon et al. 2012; Cif et al. 2007) and Hallervorden-Spatz disease (Umemura et al. 2004; Timmermann et al. 2010; Ge et al. 2011) is considered to be an indication of DBS.
Prior to considering DBS, brain MRI is always required to confirm diagnosis and assess structural abnormalities. Other imaging techniques, particularly functional imaging (Chernov et al. 2008), are used for research purposes (Thobois et al. 2011). There is no enough evidence to refuse or support consideration of DBS in patients with previous ablative procedures such as pallidotomy and thalamotomy (Bronte-Stewart et al. 2011).
10.4 Targets and Localization
The sensori-motor area of the globus pallidum interna (GPi) is the gold standard target of DBS for dystonia. However, we still do not know where the best point in the sensori-motor area of GPi is. The tentative target coordinate is 2 mm anterior to mid-commissural point, 4 mm below the commissural line, and 20 mm from the midline. The laterality is usually adjusted based on the relation of the optic tract on T2-weighted images of MRI. The target should lie just above the optic tract. This target is same as Laitinen’s pallidotomy (Laitinen et al. 1992b), and it works in majority of cases, though if this point is best to control dystonia is debatable.
There are some reports that the subthalamic nucleus (STN) can be an alternative target of DBS for dystonias (Pahapill and O’Connell 2010; Kleiner-Fisman et al. 2007; Novak et al. 2008; Sun et al. 2007; Schrock et al. 2009). STN has both theoretical and clinical evidences as a target of DBS in PD. However, still the mechanisms of dystonia control are not well known. I consider that STN DBS for dystonia is not stimulating STN itself, but that fibers from internal pallidum to ventro-oral nucleus of the thalamus are stimulated at the rostral area to STN which is the lenticular fasciculus or Forel H field. This accounts for the facts that relatively low voltage stimulation is enough to control dystonic symptoms. In recent years in China, STN stimulation is more commonly performed than pallidal DBS for dystonias.
Data on thalamic DBS for dystonia are scarce. Although thalamic DBS may be effective for focal hand dystonia such as writer’s cramp (Fukaya et al. 2007), I prefer Vo thalamotomy for such writer’s and musician’s focal hand dystonia, because the symptom is usually unilateral, patients are socially active, and there is no concern of hardware-related complications (Taira et al. 2003a, b; Horisawa et al. 2013).
10.5 Surgical Procedures
Surgical procedures of DBS differ from center to center (Abosch et al. 2013), and it is difficult to determine which the best is. Some neurosurgeons use frameless system or robotic device, while others are happy with the traditional stereotactic frames. Many neurosurgeons use intraoperative electrophysiological technique, and consider it very important, while some others rely on purely image-guided technique.
General anesthesia is often used for DBS for dystonias (Abosch et al. 2013) because dystonic movements may interfere accurate fixation of the stereotactic frame, MR scanning, and operative procedures. However, in my series of DBS for dystonias, we use general anesthesia only for small children, and did not use GA for adults. The only exception was a man who had a history of severe acute allergic reaction to lidocaine. This is because of availability of anesthetists and MR-compatible anesthetic devices. However, we seldom feel necessity of GA even in dystonia DBS. In our experience, we can easily fix the stereotactic frame and align the axes of the frame to the cerebral axes only with local anesthesia without sedation, even if dystonic symptom is severe. The frame is fixed in the sitting position on a wheel chair of which back-support can be reclined (Fig. 10.1). This is usually done in the ward. We used Leksell G frame (Elekta) and its ear-bars to align the frame. Fixing the frame takes about 10 min and then the patient is sent for MR scanning.

Fig. 10.1
Frame fixation in sitting position. Even if the patients head and neck move involuntary, fixation is not difficult without sedation
We use 1 mm thickness T1 axial and T2 coronal images without imaging gap or overwrap. The image data are transferred online to the Surgiplan computer (Elekta). The GPi target is chosen at the point described already. The angle of trajectory is determined not to pass through the ventricles (Fig. 10.2). Usually, the location of bur holes come 3.5–4.0 cm from the midline and just anterior to the coronal suture.
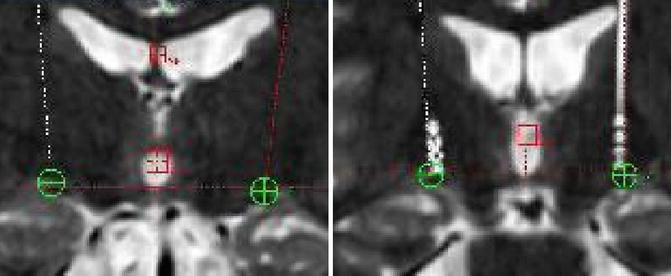
Fig. 10.2
Left: planning of GPi target. The target is just dorsal to the lateral edge of the optic tract. Right: Postoperative MR image showing accurate placement of the electrode
In the operation room, the frame is fixed with Mayfield cramp to the operation table. Care should be taken not to flex the neck excessively to avoid airway obstruction. We may use mild sedation with intravenous diazepam or propofol, but this is exceptional. C-arm X-ray fluoroscope is set, and the patient head is sterilized with alcoholic and iodine solutions (Fig. 10.3). We do not shave the head totally but local hair clipping is enough, and this does not increase the risk of infection. After appropriate draping, two burr holes are opened symmetrically with 2.5 cm straight incisions, but the dura is not opened until the last minutes of electrode insertion. Once the electrode insertion is ready, the axes of X-ray fluoroscope are well aligned in right–left direction by using cross-hairline markers (Fig. 10.4).
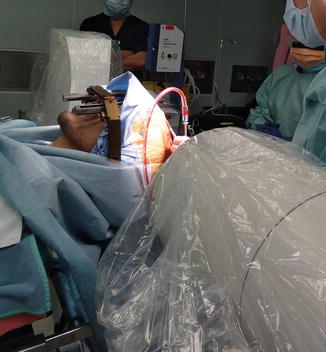

Fig. 10.3
Positioning of the patient and set-up of fluoroscope
Fig. 10.4
X-ray beam should be aligned with cross-hairline markers before opening the dura
We use a stainless-steel outer cannula (outer diameter: 2.1 mm, inner diameter: 1.5 mm, length: 20 cm) with a rigid inner stylet of 1.3 mm in diameter for DBS electrode placement. The stylet protrudes about 1.5 cm from the tip of the outer cannula. Once the tip of the stylet reaches to the target, the stylet is replaced by DBS electrode (Model 3387, Medtronic) and then trial macro-stimulation is started. Initially, we usually use 0-1+ contacts with 130 Hz, 200 μs pulse width. The voltage is gradually increased until pyramidal symptoms appear. Usually this threshold is 4.0–4.5 V. For such intraoperative macrostimulation, we use a gas-sterilized DBS screening device (Fig. 10.5). This is very useful and handy, and we have been using it for more than 10 years and have not encountered dysfunction of the device despite gas sterilization. If pyramidal symptoms appear at voltage lower than 3.0, we check the contact 1-2+. If this combination of contacts shows lower threshold than 3.0 V, the trajectory is too close to the pyramidal tract, and we move the target 2 mm more laterally. Once the electrode is appropriately placed, we remove the outer cannula carefully so that the tip of the DBS electrode does not move. Then the DBS electrode is fixed to the skull with a titanium plate coved with a silastic tube (Fig. 10.6). A small piece of surgical is placed in the burr hole. We used to tunnel out the electrode for trial stimulation, but nowadays we just place the covered proximal end of the DBS electrode subcutaneously by making a separate small incision in the posterior parietal region. We can implant the pulse generator (IPG) immediately after placing DBS electrode, but again because of availability of anesthetists, we implant IPG about a week later. After surgery, the patient is always sent for MR and CT scanning to check the location of the electrode. Fusion images of both postoperative MR and CT is useful to identify the actual location of every contact of the DBS electrode (Fig. 10.7). With the above technique, it take about less than 2 h for bilateral pallidal DBS from skin to skin, and 5 h from putting frame on and removing frame off.

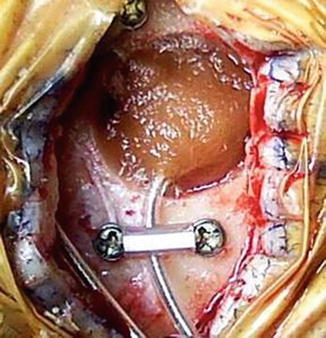
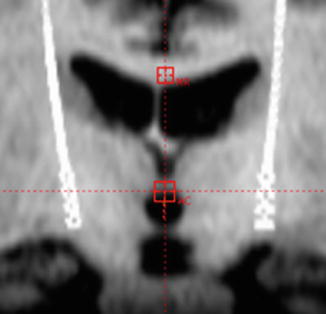
Fig. 10.5
Stimulation device for intraoperative test stimulation. This is gas-sterilized and controlled by the surgeon
Fig. 10.6
Fixation of the DBS electrode to the skull. The mini-plate is covered with a silastic tube. If necessary, we may insert two or three DBS leads from a burr hole, and they can be fixed with this method
Fig. 10.7
 Deep Brain Stimulation for Psychiatric Disorders
Deep Brain Stimulation for Psychiatric Disorders
 Symptoms and Signs of Parkinson’s Disease and Other Movement Disorders
Symptoms and Signs of Parkinson’s Disease and Other Movement Disorders
 Deep Brain Stimulation for Epilepsy: Evidence for Limbic Network Disruption via Chronic Anterior Nucleus of the Thalamus and Hippocampus Stimulation
Deep Brain Stimulation for Epilepsy: Evidence for Limbic Network Disruption via Chronic Anterior Nucleus of the Thalamus and Hippocampus Stimulation
 Intraoperative Microelectrode Recording
Intraoperative Microelectrode Recording
 Deep Brain Stimulation for Essential Tremor
Deep Brain Stimulation for Essential Tremor
 Surgical Technique of Brain Stimulation
Surgical Technique of Brain Stimulation




Postoperative fusion image of MR and CT to identify the location of each contact. AC level of anterior commissure, MR midline reference
Related posts:
Deep Brain Stimulation for Psychiatric Disorders
Symptoms and Signs of Parkinson’s Disease and Other Movement Disorders
Deep Brain Stimulation for Epilepsy: Evidence for Limbic Network Disruption via Chronic Anterior Nucleus of the Thalamus and Hippocampus Stimulation
Intraoperative Microelectrode Recording
Surgical Technique of Brain Stimulation
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
