Chapter 60 Developmental Disorders of the Nervous System
Embryological and Fetal Development of the Nervous System
Mitotic Proliferation of Neuroblasts (Neuronogenesis)
Programmed Cell Death (Apoptosis)
Fissures and Sulci of Cortical Structures
Electrical Polarity of the Cell Membrane
Biosynthesis of Neurotransmitters
Cajal-Retzius Neurons of the Fetal Brain
Suprasegmental Influences on Muscle Maturation
Etiology of Central Nervous System Malformations
Molecular Genetic Classification of Malformations of the Nervous System
Clinical Expression of Selected Malformations of the Nervous System
Embryological and Fetal Development of the Nervous System
Table 60.1 shows known genetic loci and mutations in human central nervous system (CNS) malformations. In most cases, mutations affect the genetic programming of the spatial and temporal sequences of developmental processes. These range from early processes that establish the axes of the neural tube and gradients of genetic expression, to late processes that establish the identity of specific types of neurons, the type of neurotransmitter they synthesize, and the synaptic connections they make. The role of homeobox genes in the differentiation of neural structures is an aspect of development recognized relatively recently. Molecular genetic data are rapidly becoming available because of intense interest in this key to understanding neuroembryology in general and neural induction in particular (Sarnat and Menkes, 2000). Other aspects of current investigative interest include the roles of neurotropic factors, hormones, ion channels, and neurotransmitter systems in fetal brain development. Genetic manipulation in animals has created many genetic models of human cerebral malformations. These contribute greatly to our understanding of human dysgeneses and provide insights into the pathogenesis of epilepsy and other functional results of dysgeneses (Chevassus-au-Louis et al., 1999).
Malformations of the nervous system are unique. No two individual cases are identical, even when categorized as the same anatomical malformation, such as alobar holoprosencephaly, syndromic or isolated agenesis of the corpus callosum, and types 1 and 2 lissencephaly. Functional expression of anatomically similar cases also may vary widely. For example, two cases of holoprosencephaly with nearly identical imaging findings and similar histological patterns of cortical architecture and subcortical heterotopia at autopsy may differ in that one infant may have epilepsy refractory to pharmacological control, whereas the other may have no clinical seizures at all. The difference may be at the level of synaptic organization and the relative maturation of afferent input and neuronal maturation (Sarnat and Born, 1999; Sarnat et al., 2010a). A discussion of the critical sequence of events in neural maturation follows.
Neurulation
Neurulation refers to the formation and closure of the neural tube. The formation of the neural tube from the neural placode starts with the establishment of the axis in the neural plate. The three early axes—longitudinal, horizontal, and vertical—persist during life and correspond to the basic body plan of all vertebrates (Sarnat and Flores-Sarnat, 2001b). At neurulation, grooving of the neural placode occurs in the anteroposterior axis. Subsequently, closure of the lateral margins of the folding neural placode ensues in the dorsal midline to form the neural tube. To accomplish closure, intercellular filaments interdigit cells of the two sides to form a veil at midline closure points and the neuropores. At this time, the neural crest separates bilaterally at the two fusing lips of the closing neural tube, and its cells migrate along predetermined pathways to form the peripheral nervous system, chromaffin tissue, melanocytes, and various other cells. Neural crest cells terminally differentiate only after reaching their final destination. The inhibitory function of versican, a chondroitin sulfate proteoglycan, is an important factor of the extracellular matrix for neural crest cell migration (Dutt et al., 2006).
Disorders of Neurulation (1 to 4 Weeks’ Gestation)
Anencephaly (aprosencephaly with open cranium) is a failure of the anterior neuropore to close at 24 days’ gestation, or perhaps to remain closed. The lamina terminalis and its derivatives fail to form, and most forebrain structures do not develop. Structures derived from the ventral part of the lamina terminalis, the basal telencephalic nuclei, may form imperfectly. Because the deficient forebrain neuroectoderm does not induce development of the overlying mesoderm, the cranium, meninges, and scalp do not close in the sagittal midline, exposing the remaining brain tissue to the surrounding amniotic fluid throughout gestation. The original induction failure, however, is probably that of mesodermal tissue on neuroectoderm and due to a defective rostral end of the notochord. Failure of craniofacial induction by the neural tube, mediated through the prosencephalic and mesencephalic neural crest, is another major pathogenetic factor (Sarnat and Flores-Sarnat, 2005).
Mitotic Proliferation of Neuroblasts (Neuronogenesis)
After formation of the neural tube, proliferation of neuroepithelial cells in the ventricular zone associated with mitoses at the ventricular surface generates neurons and glial cells. The rate of division is greatest during the early first trimester in the spinal cord and brainstem and during the late first and early second trimester in the forebrain. Within the ventricular zone of the human fetal telencephalon, 33 mitotic cycles provide the total number of neurons required for the mature cerebral cortex. Most mitotic activity in the neuroepithelium occurs at the ventricular surface, and the orientation of the mitotic spindle determines the subsequent immediate fate of the daughter cells. If the cleavage plane is perpendicular to the ventricular surface, the two daughter cells become equal neuroepithelial cells preparing for further mitosis. If, however, the cleavage is parallel to the ventricular surface, the two daughter cells are unequal (asymmetrical cleavage). In that case, the one at the ventricular surface becomes another neuroepithelial cell, whereas the one away from the ventricular surface separates from its ventricular attachment and becomes a postmitotic neuroblast ready to migrate to the cortical plate. Furthermore, the products of two genes that determine cell fate, called numb and notch, are on different sides of the neuroepithelial cell. Therefore, with symmetrical cleavages, both daughter cells receive the same amount of each, but with asymmetrical cleavage, the cells receive unequal ratios of each, which also influences their subsequent development (Mione et al., 1997). The orientation of the mitotic spindle requires centractin.
Active mitoses cease well before the time of birth in most parts of the human nervous system, but a few sites retain a potential for postnatal mitoses of neuroblasts. One recognized site is the periventricular region of the cerebral hemispheres (Kendler and Golden, 1996). Another is the external granular layer of the cerebellar cortex, where occasional mitoses persist until 1 year of age. Postnatal regeneration of these neurons after destruction of most by irradiation or cytotoxic drugs occurs in animals and may occur in humans as well. Primary olfactory receptor neurons also retain a potential for regeneration. In fact, if a constant turnover of these neurons did not occur throughout life, the individual would become anosmic after a few upper respiratory infections, which transiently denude the intranasal epithelium. A population of “stem cells” with mitotic potential in the subventricular zone and hippocampal dentate gyrus is reported (Johansson et al., 1999). These have generated considerable interest because of a potential for regeneration of the damaged adult brain and because they may be induced to mature as neurons (Schuldiner et al., 2001).
Disorders of Neuronogenesis
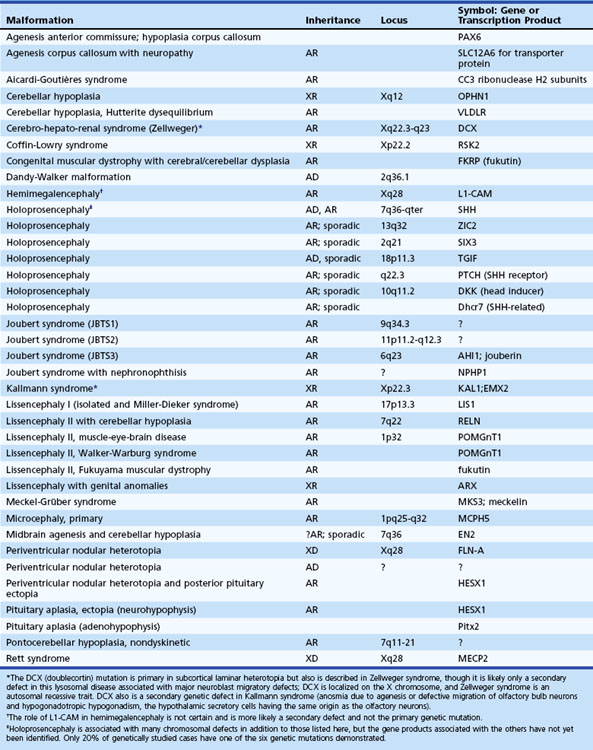
Destructive processes may destroy so many neuroblasts that regeneration of the full complement of cells is impossible. This happens when the insult persists for a long time or is repetitive, destroying each subsequent generation of dividing cells. Inadequate mitotic proliferation of neuroblasts results in hypoplasia of the brain (Fig. 60.1). Such brains are small and grossly malformed, either because of a direct effect on neuroblast migration or by destruction of the glial cells with radial processes that guide migrating nerve cells. The entire brain may be affected, or portions may be selectively involved. Cerebellar hypoplasia often is a selective interference with proliferation of the external granular layer. In some cases, cerebral hypoplasia and microcephaly are the result of precocious development of the ependyma before all mitotic cycles of the neuroepithelium are complete, because ependymal differentiation arrests mitotic activity at the ventricular surface. The mutation of a gene that programs neuronogenesis may be another explanation for generating insufficient neuroepithelial cells, although this pathogenesis remains hypothetical in humans.
Programmed Cell Death (Apoptosis)
Disorders of Programmed Cell Death
Spinal muscular atrophy (see Chapter 74) is an example of a human disease caused by apoptosis not stopping at the proper time. In this disorder, continued loss of spinal motor neurons (SMN) after the normal deletion of surplus embryonic neuroblasts expresses itself as a progressive denervating process. Genetic factors are crucial in determining the arrest of cell death, which accounts for the hereditary character of spinal muscular atrophy. The SMN defective gene at the chromosome 5q13.1 locus has now been isolated and is normally responsible for arresting apoptosis in motor neuroblasts (Roy et al., 1995).
Other neurodegenerative diseases of fetal life and infancy are more widespread within the CNS, rather than limited to one type of neuron such as the motor neuron. The characteristic feature is also progressive neuronal loss that is apoptotic rather than necrotic in character: No inflammatory or glial reaction occurs, and the features of the DNA degradation differ from ischemic necrosis. An example is pontocerebellar hypoplasia, a group of progressive degenerative diseases that begin prenatally and continue postnatally (Barth et al., 1995). Despite the name, they involve much more than the cerebellar system. These diseases are associated with extensive cerebral cortical and basal ganglionic abnormalities even in motor neurons, which cause a clinical presentation at birth resembling spinal muscular atrophy. This autosomal recessive group of diseases, all genetically distinct from olivopontocerebellar atrophy, exemplifies a semantic difficulty. If an atrophic process begins before development is complete, it results in both hypoplasia and superimposed atrophy. In the CNS, glial cells also undergo apoptosis. Glial necrosis intimately links to the interhemispheric passage of commissural fibers in the corpus callosum. In a murine model of callosal agenesis, glial cells that do not degenerate act as a barrier to crossing axons and prevent the corpus callosum from forming.
Neuroblast Migration
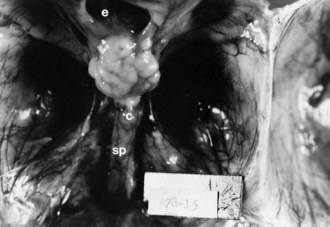
No neurons of the mature human brain occupy their site of generation from the neuroepithelium. They migrate to their mature site to establish the proper synaptic connections with appropriate neighboring neurons and send their axons in short or long trajectories to targets. The subependymal germinal matrix (Fig. 60.2) is the subventricular zone of the embryonic concentric layers and consists of postmitotic premigratory neuroblasts and glioblasts. In general, the movement of maturing nerve cells is centrifugal, radiating toward the surface of the brain. The cerebellar cortex is exceptional in that external granule cells first spread over the surface of the cerebellum and then migrate into the folia. Migration of neuroblasts begins at about 6 weeks’ gestation in the human cerebrum and is not completed until at least 34 weeks of fetal life, although the majority of germinal matrix cells after midgestation are glioblasts. Glioblasts continue to migrate until early in the postnatal period. Within the brainstem, neuroblast migration is complete by 2 months’ gestation. Cerebellar external granule cells continue migrating throughout the first year of life.
Neuroblast migration permits a three-dimensional spatial relationship to develop between neurons, which facilitates the formation of complex synaptic circuits. The timing and sequence of successive waves of migrating neuroblasts are precise. In the cerebral cortex, immature nerve cells reach the pial surface and then form deeper layers as more recent arrivals replace their position at the surface. Neurons forming the most superficial layers of neocortex are thus the last to have migrated, although in the three-layered hippocampus, the most superficial neurons represent the earliest migratory wave. Three major groups of molecules control neuroblast migration (Gressens, 2006): (1) molecules of the cytoskeleton that determine the initiation (filamin-A and ADP-ribosylation factor GEF2) and ongoing progression (doublecortin and LIS1) of neuroblast movement; (2) signaling molecules involved in lamination, including reelin and other proteins not yet associated with human diseases; and (3) molecules modulating glycosylation that provide stop signals to migrating neuroblasts (e.g., POMT1 [protein O-mannosyl-transferase], involved in Walker-Warburg syndrome; POMGnT1 [protein O-mannose β-1,2-N-acetylglucosaminyltransferase], involved in muscle-eye-brain disease; and fukutin, involved in Fukuyama muscular dystrophy).
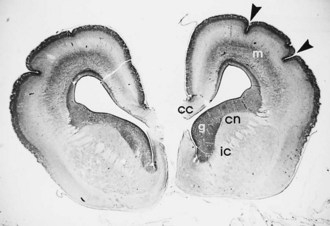
The laminated arrangement of the mammalian cerebral cortex requires a large cortical surface area to accommodate increasing numbers of migrating neuroblasts and glioblasts. Initially the cortical plate shows no histological layering, a process beginning at about midgestation, but rather has an immature columnar architecture. The lamination is superimposed upon this columnar pattern, but columnar architecture is still seen postnatally, particularly at the crowns of gyri and the depths of sulci. Even before histological lamination is evident, RNA probes for specific neuronal identities can already detect future organization of the cortical plate (Hevner, 2007). Convolutions provide this large surface area without incurring a concomitant increase in cerebral volume. The formation of gyri and sulci is thus a direct result of migration (Fig. 60.3). Most gyri form in the second half of gestation, which is a period of predominant gliogenesis and glial cell migration. Therefore, the proliferation of glia in the cortex and subcortical white matter may be more important than neuroblast migrations in the formation of convolutions, but the growth of dendrites and synaptogenesis also may influence gyration by contributing mass to the neuropil.
Major Mechanisms of Neuroblast Migration: Radial Glial Fiber Guides and Tangential Migration Along Axons
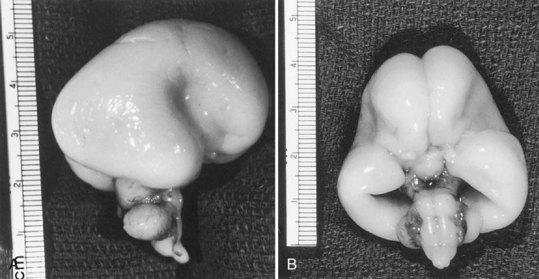
The majority of neuroblasts arriving at the cortical plate do so by means of radial glial guides from the subventricular zone. A second route, tangential migration, uses axons as the guides for the migratory neuroblasts. The genetically determined programming of neuroblast migration begins when cells are still undifferentiated neuroepithelial cells and even before all their mitotic cycles are complete. Neuroepithelial cells express the gene products of the lissencephaly gene (LIS1), as do ependymal cells and Cajal-Retzius cells of the molecular layer of cerebral cortex. The expression of this gene is defective in type 1 lissencephaly (Miller-Dieker syndrome), a severe disorder of neuroblast migration (Clark et al., 1997). An understanding of its function in migration is incomplete. The guidance of most neurons of the forebrain to their predetermined site from the germinal matrix (embryonic subventricular zone) is by long radiating fibers of specialized fetal astrocytes (Fig. 60.4). The elongated processes of these glial cells span the entire wall of the fetal cerebral hemisphere; their cell bodies are in the periventricular region, and their terminal end-feet are on the limiting pial membrane at the surface of the brain (see Fig. 60.4). Radial glial cells are the first astroglial cells of the human nervous system converted into a mature fibrillary astrocyte of the subcortical white matter; some are still present at birth. Mature astrocytes are present throughout the CNS by 15 weeks’ gestation, and gliogenesis continues throughout fetal and postnatal life. Several types of glial cells are recognizable between 20 and 36 weeks’ gestation.
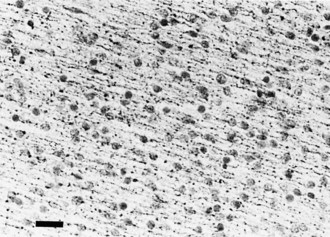
Facilitating the mechanical process of neuroblasts gliding along a radial glial fiber are several specialized proteins at the radial glial fiber surface membrane or extracellular space. An example is astrotactin, secreted by the neuroblast (Zheng et al., 1996). Glial cells and neural cell adhesion molecules also facilitate gliding (Jouet and Kenwrick, 1995). Fetal ependymal cells have radiating processes that resemble those of the radial glial cell but do not extend beyond the germinal matrix and secrete molecules in the extracellular matrix. Some adhesion molecules are present in the extracellular matrix (Thomas et al., 1996). These molecules serve as lubricants, as adhesion molecules between the membranes of the neuroblast and the radial glial fiber, and as nutritive and growth factors. They stimulate cell movement by a mechanism still poorly understood. Deficient molecules lead to defective migration. For example, the abnormality of the L1 adhesion molecule is the defective genetic program in X-linked hydrocephalus accompanied by polymicrogyria and pachygyria.
In addition to the radial migration to the cerebral cortex, tangential migration also occurs, but the number of neuroblasts is far smaller (Rakic, 1995; Takano et al., 2004). These migrations perpendicular to the radial fibers probably use axons rather than glial processes as guides for migratory neuroblasts. This explains why not all cells in a given region of cortex are from the same clone or vertical column. Most of the tangentially migrating neuroblasts in the cerebral cortical plate are generated in the fetal ganglionic eminence, a deep telencephalic structure of the germinal matrix that gives origin to basal ganglionic neurons and to the γ-aminobutyric acid (GABA)ergic inhibitory interneurons of the cerebral cortex. These neurons in the cortex from tangential migration have some unique metabolic features such as calretinin synthesis (Takano et al., 2004; Ulfig, 2002). Calretinin-reactive inhibitory interneurons in the cerebral cortex comprise about 12% of total neurons and are a subset of total neurons arriving at the cortical plate by tangential migration, which represent about 20% of total cortical neurons.
Tangential migrations occur in the brainstem and olfactory bulb as well as in the cerebrum. The subpial region is another site of neuroblast migration that does not use radial glial cells. Calretinin-reactive neurons are in the cerebellum as well as the cerebral cortex (Yew et al., 1997), particularly Purkinje cells, basket cells, and neurons of the dentate and inferior olivary nuclei of the cerebellar system, but not those of the pontine nuclei, which similarly originated in the rhombic lip of His.
Disorders of Neuroblast Migration
Most disturbances of neuroblast migration involve arrested migration before the journey is complete. These disorders are divisible into three anatomical phases, depending on where the migratory arrest occurred. An example of neuroblasts never having begun migration from the periventricular region is periventricular nodular heterotopia, an X-linked genetic disorder due to defective expression of the gene, filamin-A (FLNA). Subcortical laminar heterotopia results when neuroblasts begin migration but arrest in the subcortical white matter before reaching the cortical plate. This is another X-linked recessive trait but is due to a different gene called doublecortin (DCX). The term double cortex is sometimes used, but this name is incorrect because unlike a true cortex, the subcortical heterotopia lacks lamination. If the neuroblasts reach the cortical plate but lack correct lamination, accompanying this abnormal architecture of the cortical plate are abnormalities of gyration such as lissencephaly or pachygyria. Several different genes, including LIS1 and reelin (RLN), are important in cortical plate organization (Curran and D’Arcangelo, 1998) and mutated in malformations of the terminal phase of neuroblast migration.
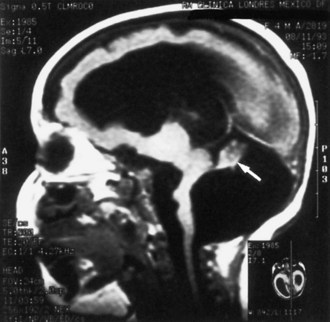
Lissencephaly is a condition of a smooth cerebral cortex without convolutions. Normally at midgestation, the brain is essentially smooth; the interhemispheric, sylvian, and calcarine fissures are the only ones formed. Gyri and sulci develop between 20 and 36 weeks’ gestation, and the mature pattern of gyration is evident at term, although some parts of the cerebral cortex (e.g., frontal lobes) are still relatively small. In lissencephaly type 1 (Miller-Dieker syndrome), the cerebral cortex remains smooth. Lesser degrees of this gross morphological defect exist, with a few excessively wide gyri (pachygyria) or multiple excessively small gyri (polymicrogyria). The histopathological pattern is that of a 4-layer cortex in which the outermost layer (1) is the molecular layer, as in normal 6-layered neocortex. Layer 2 corresponds to layers 2 through 6 of normal neocortex, layer 3 is cell-sparse as a persistent fetal subplate zone, and layer 4 consists of incompletely migrated neurons in the subcortical intermediate zone. In lissencephaly type 2 (Walker-Warburg syndrome), poorly laminated cortex with disorganized and disoriented neurons is seen histologically, and the gross appearance of the cerebrum is one of a smooth brain or a few poorly formed sulci (Fig. 60.5). The term cobblestone refers to the aspect of the surface with multiple shallow furrows not corresponding to normal sulci. The cerebral mantle may be thin, suggesting a disturbance of cell proliferation as well as of neuroblast migration. Malformations of the brainstem and cerebellum often are present as well (see Fig. 60.5). Lissencephaly type 1 and type 2 (Walker-Warburg syndrome, Fukuyama muscular dystrophy, muscle-eye-brain disease of Santavuori) are genetic diseases. Lissencephaly also results from nongenetic disturbances of neuroepithelial proliferation or neuroblast migration, including destructive encephaloclastic processes such as congenital infections during fetal life. More recently it has been recognized that the lissencephalies, including those resulting from mutations in LIS1, DCX, and ARX genes, are disturbances not only in radial migration, but also involve tangentially migrating neuroblasts (Marcorelles et al., 2010).
In sum, either defective genetic programming or acquired lesions in the fetal brain that destroy or interrupt radial glial fibers may cause disorders of neuroblast migration. Cells may not migrate at all and become mature neurons in the periventricular region, as occurs in X-linked periventricular nodular heterotopia (Eksioglu et al., 1996) and in some cases of congenital cytomegalovirus infection. Cells may become arrested along their course as heterotopic neurons in deep subcortical white matter, as occurs in many genetic syndromes of lissencephaly-pachygyria and in many metabolic diseases including cerebrohepatorenal (Zellweger) syndrome and many aminoacidurias and organic acidurias. The same aberration may occur in acquired insults to the radial glial cell during ontogenesis. Cells may overmigrate beyond the limits of the pial membrane into the meninges as ectopic neurons, either singly or in clusters known as marginal glioneuronal heterotopia, or brain warts. Rarely, herniation of the germinal matrix into the lateral ventricle may occur through gaps in the ependyma; those cells mature as neurons, forming a non-neoplastic intraventricular mass that may or may not obstruct cerebrospinal fluid (CSF) flow. Whether disoriented radial glial fibers actually guide neuroblasts to an intraventricular site or neuroblasts are physically pushed in a direction of less resistance is uncertain.
Fissures and Sulci of Cortical Structures
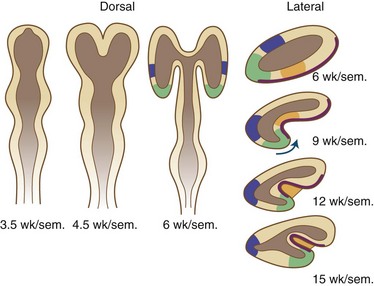
Fissures and sulci are grooves that form in laminated cortices, principally cerebral and cerebellar. Such folding accomplishes a need for an enlarging surface area without a concomitant increase in tissue volume as development proceeds. Without gyration of the cerebral cortex and foliation of the cerebellar cortex, the brain would be so large and voluminous at birth that neither the neonate nor the mother would survive delivery. Fissures and sulci both result from mechanical forces during fetal growth, but they differ in that fissures form from external forces and sulci form from internal forces imposed by the increased volume of neuronal cytoplasm and the formation of neuropil, the processes of neurons and glial cells (Sarnat and Flores-Sarnat 2010a). The ventricular system acts as another external force, surrounded by but outside of the brain parenchyma. Whereas fissures generally form earlier and often are deeper than sulci, these are not the most important differences. Box 60.1 lists the various fissures of the brain, and Fig. 60.6 is a drawing of the development of the human telencephalic flexure, which becomes, after closure of the operculum, the Sylvian fissure. It should be noted that the ventral bending of the primitive oval-shaped telencephalic hemisphere results in the original posterior pole becoming the temporal—not the occipital—lobe, and that the lateral ventricle bends with the brain. The occipital horn of the lateral ventricle is a more recent diverticulum of the original simple ventricle and as such remains the most variable part of the ventricular system, symmetrical in only 25% of normal individuals. Cerebellar folia are the equivalent of cerebral cortical gyri. A temporally and spatially precise sequence of the development of fissures, sulci, and cerebellar folia is genetically programmed and enables the neuroradiologist and neuropathologist to also assess maturational delay of this aspect of ontogenesis. The gestational age of a premature infant may be determined to within a 2-week period or less from the convolutional pattern of the brain.
Disorders of Fissures and Sulci
The telencephalic Sylvian fissures fail to form in holoprosencephaly and form abnormally in many major malformations of the brain, including lissencephalies, schizencephaly, and severe cerebral hypoplasias (Sarnat and Flores-Sarnat, 2010a). Abnormal gyration is a regular feature of many neuroblast migratory disorders, including lissencephaly, pachygyria, and polymicrogyria, enabling an accurate diagnosis by neuroimaging not only postnatally but also by prenatal fetal magnetic resonance imaging (MRI), even though microscopic details of cortical lamination and organization are below the resolution of these techniques.
Growth of Axons and Dendrites
< div class='tao-gold-member'>
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


