Chapter 24 Disorders of Cerebellar and Brainstem Development
A number of newly described syndromes and diseases now allow for careful consideration of cerebellar and brainstem malformations in children. Because computed tomography (CT) scanning offers poor resolving power of the hindbrain due to tight encasement in the posterior fossa, the complete description of these diseases has lagged behind those involving the cerebral cortex. The introduction of magnetic resonance imaging (MRI) in the last 20 years has provided for the delineation of new syndromes, many of which were previously lumped together as “Dandy–Walker variants” (Table 24-1).
Chiari I Malformation
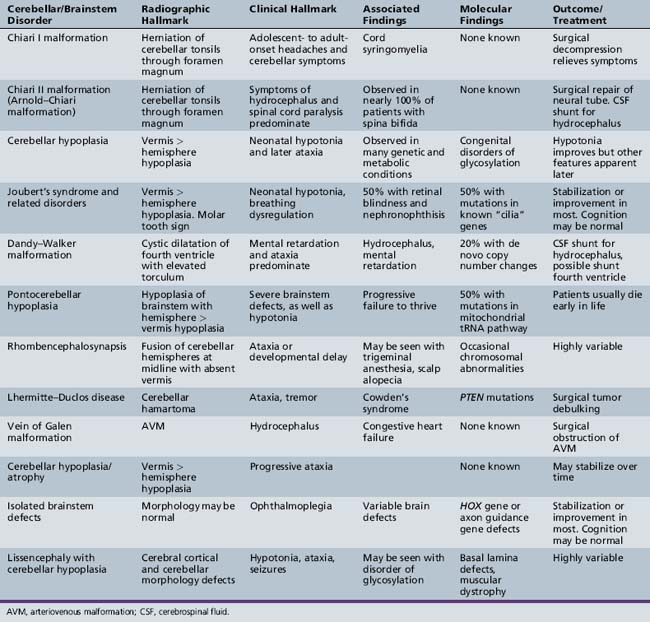
The Chiari I malformation is a disorder of uncertain origin that traditionally has been defined as a downward herniation of the cerebellar tonsils of at least 3–5 mm through the foramen magnum [Tubbs et al., 2007] (Figure 24-1). The anomaly is a leading cause of syringomyelia and occurs in association with osseous abnormalities of the craniovertebral junction. It must be distinguished from the Chiari II malformation (also known as Arnold–Chiari malformation) and the rare Chiari III malformation.
Pathophysiology
Although a number of theories as to the etiology of the Chiari I malformation have been suggested, no clear consensus has emerged. One popular theory that was dispelled was the idea that tethering of the spinal cord causes posterior fossa structures to be pulled into the spinal canal. Another theory gaining favor is that congenital hypoplasia of the posterior fossa occurs due to disrupted mesodermal development [Marin-Padilla and Marin-Padilla, 1981]. There is an increased risk of developing this malformation with growth hormone deficiency and bone defects like rickets. Female patients greatly outnumber male patients, and there is familial clustering, suggesting a genetic predisposition. Gene mutations of chromosomes 9 and 15 may contribute to development of this malformation [Boyles et al., 2006], and it also has recently been linked to Ehlers–Danlos syndrome [Castori et al., 2010]. In about 20 percent of individuals, there is identified trauma that precedes symptom onset.
Clinical Characteristics
Initial clinical features largely depend on whether syringomyelia is present, since this defect tends to dominate the clinical picture. Symptoms from syringomyelia, present in 30–70 percent of adult Chiari I patients but in only 12 percent of children, depend on the location and size of the syrinx and can occur as a result of stretching and distention of the cord [Milhorat et al., 1999; Aitken et al., 2009]. Long-tract findings include hyperreflexia, urinary incontinence, muscle wasting, loss of proprioception, and arm numbness/tingling.
Management
Surgical decompression of the posterior fossa with duraplasty is the treatment of choice in selected cases. The purpose of this surgery is to enlarge the bony compartment. It is important to evaluate for coexistent hydrocephalus before any correction is attempted. In patients with a coexistent syrinx, there is a clear indication for surgery, and in a series of 49 children with Chiari-associated syringomyelia, just over half demonstrated clinical and radiographic improvement after hindbrain decompression [Attenello et al., 2008]. In these patients, decompression of the posterior fossa alone is often sufficient to reduce the syringomyelia without directly shunting the fluid collection. In patients without a syrinx but with >5 mm of caudal displacement or with progressive features, surgery may be offered. In some individuals, clinical symptoms and herniation may spontaneously improve, so a conservative approach is generally adopted in minimally affected patients [Novegno et al., 2008] and in those with <3 mm displacement.
Cerebellar Hypoplasia
Cerebellar hypoplasia is generally an underappreciated entity until observed on brain MRI, and even then it may be missed, unless specifically looked for based upon symptomatology. Hypoplasia, as opposed to atrophy, excludes conditions in which cerebellar development was normal but then shows progressive degeneration, as in Friedreich’s ataxia, vitamin E ataxia, and ataxia with oculomotor apraxia. In patients with cerebellar hypoplasia, psychomotor delay is present in 70–85 percent, and epilepsy in 28 percent [Ventura et al., 2006], although the exact mechanism of the hypoplasia, when identified, can be a better predictor of outcome.
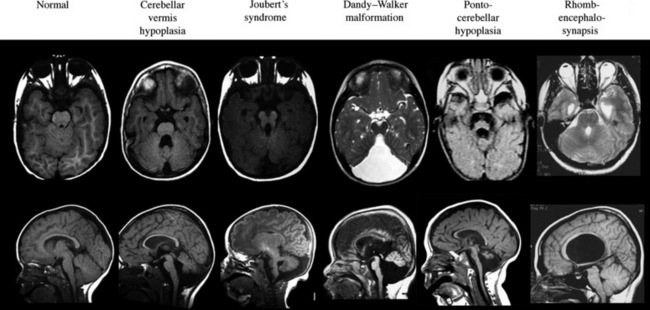
The term “global cerebellar hypoplasia” refers to a cerebellum of reduced volume (Figure 24-2). Typically, the vermis (cerebellar midline) is more severely affected than the hemispheres because the vermis is the last part of the cerebellum to form, and thus, it is probably more sensitive to alterations in genetic programs. The subarachnoid spaces may be passively enlarged. Typical causes include various chromosomal aberrations, such as trisomy 9, 13, and 18, several metabolic disorders including congenital disorders of glycosylation (CDGs) [Vermeer et al., 2007], prenatal teratogenic drugs (e.g., anticonvulsants), fetal infections, or isolated genetic cerebellar hypoplasia, such as that caused by mutations in the OPHN1 gene [Tentler et al., 1999]. Cerebellar hypoplasia can also be observed as part of other brain malformation syndromes, such as lissencephaly, pontocerebellar hypoplasia, or those associated with congenital muscular dystrophies.
Pathophysiology
The etiological basis of the cerebellar disorder predicts the pathophysiology, but generally follows some defect in cellular proliferation or differentiation of either the cerebellar Purkinje or granule cells. In the CDGs, as a result of insufficient protein glycosylation, there is probably an activation of the unfolded protein response, leading to an effect on the cerebellum (see Chapter 35). Likewise, teratogenic exposure to alcohol in animal models can affect neuronal migration by altering established pathways [Jiang et al., 2008].
Differential Diagnosis
It is critical to evaluate individuals with cerebellar hypoplasia for syndromic forms of this malformation, as well as to consider the various genetic etiologies (Table 24-2). Like most developmental brain anomalies, fetal infections, and toxic exposures, recessive and dominant inherited forms, as well as alterations in genetic copy number (using comparative genomic hybridization), should be considered. Normal transferrin isoelectric focusing can help exclude CDGs. Normal family history may help exclude genetic causes. Baseline laboratory tests, including creatine phosphokinase (CPK), vitamin E levels, and TORCH (toxoplasmosis, other infections, rubella, cytomegalovirus, and herpes simplex virus) titers, looking for prenatal viral infection, can help exclude muscle-eye-brain disease, vitamin E deficiency, and congenital infections. Mitochondrial disorders should be considered, especially in the setting of features such as growth retardation, hearing loss, acidosis, and respiratory or multi-organ involvement.
Table 24-2 Causes of Cerebellar Hypoplasia
| Cause | Gene | Reference |
|---|---|---|
| Cerebellar ataxia, mental retardation and dysequilibrium syndrome | VLDLR, CA8 | Boycott et al. [2005]; Turkmen et al. [2009] |
| Cerebellar hypoplasia with pancytopenia | DKC1 | Pearson et al. [2008] |
| Cerebellar hypoplasia, Norman type (SCAR2) recessive | Unknown | Megarbane et al. [1999] |
| Cerebellar vermis aplasia (SCA29) dominant | Unknown | Rivier and Echenne [1992] |
| Cerebellar hypoplasia, mental retardation, distinctive facies | OPHN1 | des Portes et al. [2004] |
| Cerebellar aplasia with diabetes mellitus | PTF1A | Sellick et al. [2004] |
| Congenital disorders of glycosylation | CDG1 and 2 | Jaeken and Matthijs [2007] |
Joubert’s Syndrome and Related Disorders
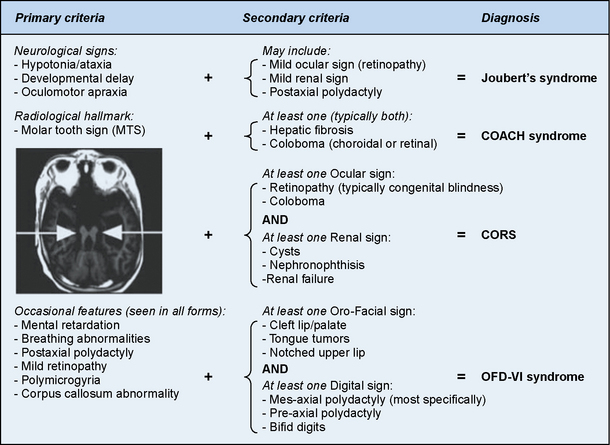
Joubert’s syndrome (JS) and related disorders (JSRDs) are a group of recessive congenital ataxia conditions, usually presenting with neonatal hypotonia, dysregulated breathing rhythms, oculomotor apraxia, and subsequently with mental retardation. JSRDs are the most common inherited congenital cerebellar malformation. The pathognomonic finding in JSRD is the unique molar tooth sign (MTS) on brain imaging (Figure 24-3). There is a tremendously broad spectrum of signs and symptoms, including kidney, retinal, and hepatic involvement, along with polydactyly and facial dysmorphisms.
Clinical Characteristics
The MTS is the result of cerebellar vermis hypoplasia, thick and maloriented superior cerebellar peduncles, and an abnormally deep interpeduncular fossa, which together give the appearance of a tooth. JS is the best-known and probably most common syndrome associated with the MTS. JS is notable for both intrafamilial and interfamilial phenotypic variability. Even in the original pedigree of four affected siblings, two had hypoplasia of the posterior inferior cerebellar vermis and two had complete agenesis of the cerebellar vermis [Joubert et al., 1968].
Clinical features are highly variable for patients with the MTS. Many patients with JS present in the newborn period with an unusual pattern of intermittent respiration, displaying alternating hyperpnea and apnea. In JS, there is no specific lung pathology to explain this respiratory abnormality. Instead, it likely relates to dysfunction of the respiratory centers located in the brainstem or cerebellum [Xu and Fraser, 2002]. Because some neonates have died from apnea, it is critical to institute careful respiratory care until this feature of JS passes, usually by 1 year of age. Some patients will have relatively mild disease, with congenital ataxia that lessens with age, and a delay in the ability to walk until age 4–5 years, but are otherwise healthy. Other patients will display congenital blindness and renal failure requiring dialysis or renal transplantation. It is important to differentiate the type of JSRD at the time of presentation, and then to monitor the patient periodically for development of any extra-central nervous system signs. If patients do not develop retinal or renal involvement by age 10–15 years, they are unlikely to do so.
The disorders related to JS include cerebellar vermis hypo/aplasia-oligophrenia-ataxia-ocular coloboma-hepatic fibrosis (COACH syndrome), cerebello-oculo-renal syndrome (CORS), and oro-facio-digital syndrome type VI (OFD-VI), all of which share the MTS, but additionally display features that distinguish them from JS [Zaki et al., 2008]. Diagnostic criteria can help distinguish each of the four major clinical entities within the JSRD spectrum.
Pathophysiology
JSRDs fall within the “ciliopathy” spectrum of conditions, which are due to alterations in development or signaling within the cellular primary cilium. Motile cilia are well known for their roles in certain tissues, such as the sperm, where they propel cells, but there is another class of cilia, termed primary cilia, which are nonmotile, typically lacking the central pair of microtubules and outer dynein arms necessary for movement. Several genes associated with JS have now been established, and protein products of these genes are found specifically at the primary cilium in most cells throughout the body. Thus, JS is now included with other ciliopathies, including Bardet–Biedl syndrome, nephronophthisis, Leber congenital amaurosis, congenital hepatic fibrosis, Jeune asphyxiating thoracic dystrophy, and Ellis–van Creveld syndrome, as a disorder of the primary cilium [Tobin and Beales, 2009].
For some of these disorders, like Leber congenital amaurosis (congenital retinal blindness), the relation between primary cilia and pathogenesis is readily apparent, given the fact that the entire photoreceptor outer segment is a modified primary cilium. Within the developing cerebellum, primary cilia have been shown to be essential for reception of the cell signaling ligand sonic hedgehog, which in turn is essential for proliferation of cerebellar granule neurons [Chizhikov et al., 2007; Spassky et al., 2008]. Thus, it is postulated that reduced granule neuron production is the central aspect of the cerebellar hypoplasia.
Differential Diagnosis
JSRDs can be distinguished by the presence of the MTS, which can be detected prenatally and used for diagnosis [Saleem and Zaki, 2010]. In the absence of the MTS, other cerebellar malformations must be considered. In some families, there can be one child with JS and a second child with either a more severe disorder like Meckel–Gruber syndrome, or a less severe disease like Bardet–Biedl syndrome. This heterogeneity cannot be clearly explained by genetics alone, since even identical twins can show divergent severity [Raynes et al., 1999]. It is important to evaluate the whole family to determine if children with such divergent disease might share the same underlying genetic cause.
Management
The management of JSRD can be aided by anticipation of typical problems and the use of genetic testing to help determine prognosis. Patients identified in the neonatal intensive care unit should be monitored closely for apnea. Treatment with caffeine may help promote respiratory drive and an apnea monitor may be required for the first year of life. Because of the heterogeneity of these conditions, it is recommended that patients be evaluated by a geneticist and undergo screening for coexisting medical problems, including brain MRI, polysomnogram, echocardiogram, careful dysmorphology and retinal evaluation, and annual monitoring with liver function testing and renal ultrasound examinations [Parisi and Dobyns, 2003].
Prenatal diagnosis may be approached in one of several ways. JS is one of the disorders that may be encountered when cerebellar hypoplasia is detected on prenatal ultrasound. In this case, an ultrasonographer may observe nonspecific hypoplasia, and then will seek fetal brain MRI for more specific diagnosis. JS may diagnosed on brain MRI, based on the presence of the MTS [Saleem and Zaki, 2010]. Alternatively, prenatal diagnosis can be made genetically, especially in certain populations with founder mutations [Valente et al., 2010].
Genetic testing can predict disease severity, based upon established genotype–phenotype correlations. Mutations in certain genes for JS, such as AHI1, INPP5E, CC2D2A, and ARL13B, can be detected in patients with MTS who do not have involvement of other organs, or they can be detected in patients with MTS together with retinal blindness. Mutations in TMEM216 and RPGRIP1L genes are often encountered in those patients with MTS and renal involvement. Mutations in CEP290 are encountered in half of patients with MTS together with retinal blindness and renal involvement. Mutations in TMEM67 are the most common cause of MTS with liver involvement [Brancati et al., 2009]. Thus, the specific gene mutation identified in a patient can help predict the range of organ involvement.
Dandy–Walker Malformation
Dandy–Walker malformation (DWM) is defined by hypoplasia and upward rotation of the cerebellar vermis, accompanied by cystic dilatation of the fourth ventricle and frequently an enlarged posterior skull. It was first described by Dandy and Blackfan in 1914, and is the most common congenital malformation of the cerebellum with an incidence of about 1 in 5000 live births [Parisi and Dobyns, 2003]. Previous definitions of DWM have included features such as the presence of hydrocephalus or the presence of communication between the posterior fossa and the fourth ventricle. Furthermore, the now outdated term, Dandy–Walker variant, used to describe all cerebellar malformations before the routine use of brain MRI, should be replaced using stricter criteria.
This triad can be found in association with supratentorial hydrocephalus, which traditionally has been considered a complication rather than a part of the malformation complex [D’Agostino et al., 1963; Hart et al., 1972]. New data, however, suggest that the hydrocephalus may reflect a common developmental disruption affecting multiple brain regions [Aldinger et al., 2009].
Clinical Characteristics
The clinical spectrum associated with DWM is extremely broad, ranging from asymptomatic to severe mental retardation. Half of the patients have normal cognition, whereas others never achieve normal intellectual function, even when hydrocephalus is treated effectively. The vermis anatomy in DWM is statistically correlated with neurological and intellectual outcome [Klein et al., 2003]. Systemic malformations associated with DWM may include cardiac anomalies (ventriculoseptal defects, patent ductus arteriosus, transposition of the great arteries), urogenital anomalies (hydroceles, vesico-ureteral reflux, abnormal kidney shape), and other abnormalities (duodenal atresia, cleft palate, malformed limbs), and occur collectively in about half of patients [Sasaki-Adams et al., 2008]. Those with isolated DWM can have normal development.
Pathophysiology
The low empiric recurrence risk of approximately 1–2 percent for nonsyndromic DWM suggested that a simple mode of inheritance was unlikely [Murray et al., 1985], and the frequent association with chromosomal anomalies supported this hypothesis. Accumulating evidence suggests that many cases represent de novo alterations in genomic integrity, with focus on chromosome 3q2 and 6p25.3 associated with the Axenfeld–Rieger syndrome [Aldinger et al., 2009]. Additionally, a dominant form has been linked to chromosome 2q36.1 in association with occipital encephalocele.
The genes ZIC1, ZIC4, and FOXC1 when deleted can result in DWM. The ZIC1 [Grinberg et al., 2004; Aldinger et al., 2009] and ZIC4 genes are produced in cerebellar primordial cells, whereas the FOXC1 gene is produced by the developing mesenchyme, which gives rise to the meninges and posterior skull, overlying the developing cerebellum. This suggests at least two different mechanisms of disease, one in which some regulatory genes are required in the developing cerebellum itself, and others in the adjacent mesenchyme, with DWM a result of either defect. Because all genes identified to date are transcription factors, DWM is presumably a disorder associated with altered cellular programming during development; however, further research is required to delineate these mechanisms.
Differential Diagnosis
Previous literature also used the term Dandy–Walker variant, which included any of the various cerebellar hypoplasias; this should be dropped in favor of more specific terminology. It is important to consider mega cisterna magna, retrocerebellar cysts, and Blake’s pouch cyst in the differential diagnoses [Nelson et al., 2004]. In each of these disorders, there is a fluid collection in the posterior fossa adjacent to the cerebellum, but not within the fourth ventricle, without elevation of the torcula; they thus do not meet the criteria for DWM. Additionally, in these three conditions, which are often found incidentally, the prognosis is usually excellent. In some cases, imaging findings are intermediate between one of these conditions and DWM, and expert review might be necessary to arrive at a precise diagnosis. As mentioned, the association with hydrocephalus, congenital heart disease, urogenital anomalies, and cleft lip/palate can support the diagnosis of DWM.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


