Proximal greater than distal weakness
Symmetrical weakness
Muscle atrophy proportional to degree of weakness
Muscle may feel doughy to palpation
Hypotonic muscle
Weakness slowly progresses
Weakness rarely painful
Loss of deep tendon reflexes is proportional to degree of weakness
Sensory loss does not occur
Serum creatine kinase (CK) is often elevated early in disease
Electromyography shows myopathic features
Table 4.2 lists differences between the various types of weakness that are helpful in localizing weakness to a muscle disorder.
Table 4.2
Distinguishing characteristics of limb weakness
Characteristic | Upper motor neuron (corticospinal tract) | Lower motor neuron (peripheral nerve) | Distal polyneuropathy | Neuromuscular junction | Skeletal muscle |
|---|---|---|---|---|---|
Muscle involved | Distal > proximal and often unilateral | Distal > proximal | Distal > proximal | Proximal > distal | Proximal > distal |
Muscle atrophy | Minimal | Marked | Moderate | Minimal | Moderate |
Normal strength that quickly fatigues | No | No | No | Yes | No |
Fasciculations | No | Common | No | No | No |
Deep tendon reflexes | Increased | Decreased to absent | Decreased to absent | Normal or slightly decreased | Normal to decreased proportional to weakness |
Sensory loss | May be unilateral | Yes | Yes | No | No |
Family history positive | Uncommon | Uncommon | Uncommon | Uncommon | Common |
CK elevation | No | No | No | No | Yes |
EMG findings | None | Denervation or slow motor nerve conduction velocity | Few | Few | Myopathic |
Muscle diseases are divided into 4 broad categories: muscular dystrophy due to genetic abnormalities, channelopathies with abnormal sodium, calcium or potassium membrane ion channels, inflammatory myopathies, and secondary endocrine myopathies.
Duchenne Muscular Dystrophy (Muscular Dystrophies)
Introduction
Congenital muscular dystrophies are characterized by both genetic and clinical heterogeneity. They vary in the age of onset, location of muscle involvement, and whether they affect males or females. The most common muscular dystrophy is Duchenne muscular dystrophy (DMD), which is a severe, progressive disorder. The incidence is 1 in 3500 newborn boys. The de novo (new mutation) frequency is 1 in 3 cases. DMD is a lethal disorder of childhood associated with a marked deficiency or absence of dystrophin . The largest gene in the human genome, located on the X chromosome at Xp21, encodes dystrophin . Duchenne’s muscular dystrophy is the most common disease associated with genetic mutations of the dystrophin gene. Collectively these diseases are called dystrophinopathies. As DMD is transmitted by X-linked recessive inheritance, nearly all patients are male. About 10 % of female carriers have mild muscle weakness .
Pathophysiology
The dystrophin gene is the largest known, spanning about 2.2 megabases of DNA or almost 1 % of the entire X chromosome. Muscle dystrophin is a large 427-kDa molecular weight protein of 3685 amino acids that is found primarily within skeletal, smooth, and cardiac muscles. Dystrophin isoforms are also present in cortical neurons, Purkinje cell neurons, glia, and Schwann cells . Dystrophin accounts for 5 % of sarcolemmal cytoskeletal proteins in muscle. The protein is rod shaped and resides just beneath the sarcolemmal membrane as two parallel fibers (Fig. 4.1). The amino terminus is attached to actin, and the carboxyterminus binds to a transmembrane protein complex that is transmembrane located (Fig. 4.1). In muscle, dystrophin links myofibrillar elements with the sarcolemma, affording stability and flexibility to the muscle fiber.
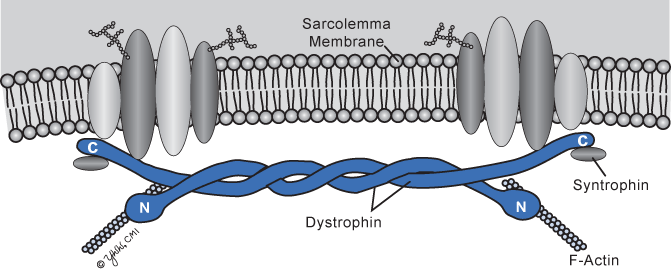
Fig. 4.1
Dystrophin molecule
Seventy-five percent of patients demonstrate large-scale deletions in the gene or have partial gene duplications, and the remaining are poorly characterized. Nearly 80 % of deletions occur in the center of the protein. The remaining 25 % of patients have small or point mutations. In DMD, the C-terminal domain is lacking and therefore the dystrophin protein is nonfunctional. In Becker muscular dystrophy (BMD), a milder form of DMD, the dystrophin protein , is internally truncated but contains the normal interacting domains—maintaining partial functionality.
Dystrophin gene mutations that cause DMD result in either the absence of dystrophin protein production or markedly truncated proteins that cannot attach to the transmembrane protein complex and are rapidly catabolized. The net result is the virtual absence of dystrophin and the DAP complexes along the sarcolemmal membrane. Quantitative studies of dystrophin have shown that less than 3 % of normal dystrophin content is present in DMD muscle (Fig. 4.2).
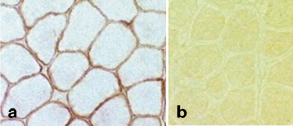
Fig. 4.2
a Normal dystrophin staining around rim of muscle fiber. b Absent dystrophin staining in Duchenne muscular dystrophy. (Courtesy of Alan Pestronk, MD)
The absence of dystrophin leads to membrane instability, myofiber leakiness of creatine kinase (CK) , and susceptibility to injury from normal muscle contractions. Over time, the damaged muscle cell wall allows abnormal influxes of calcium and activation of cell proteases with amplification of disturbed calcium homeostasis, resulting in fiber necrosis, secondary inflammation, and apoptosis.
Although mature muscle fibers are post-mitotic, skeletal muscle contains mononuclear muscle precursor cells that proliferate and fuse in response to stimuli from degenerating muscle fibers. Since these regenerating muscle fibers are affected by the gene mutation, the new muscle fibers contain the dysfunctional dystrophin and the cycle repeats. Over time, fibrosis and scarring develop in the muscle. Degenerating muscle cells are replaced by fat cells. This fatty replacement and fibrosis result in a pseudo-hypertrophic appearance of the weakened muscles.
Mutations in the DMD gene cause a spectrum of diseases of which DMD and BMD are the most severe—but also includes X-linked dilated cardiomyopathy, cramps and myalgias, and metabolic derangements such as asymptomatic hyperCKemia.
Major Clinical Features
Although children with DMD have disease activity in the neonatal period (elevated serum CK and necrosis on muscle biopsy), they rarely have clinical symptoms until age 3–4 years. However, within the first year of life, boys with DMD can have mild language delay. Parents usually report difficulty in running or climbing, frequent falls, and enlargement of the calf muscles that feel firm and rubbery. By 4–5 years of age, the gait becomes wide-based and waddling (also known as Trendelenburg gait). Affected children often walk on their toes because of contractures of heel cords. Weakness is greatest in proximal muscles and produces a compensatory Gower’s maneuver (placing hands on the knees and climbing up their thighs when trying to rise from the ground or a chair (Fig. 4.3).
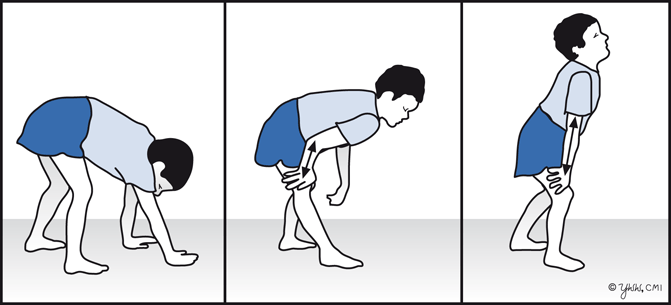
Fig. 4.3
Gower’s maneuver
In the early school years, the limb weakness progresses and is accompanied by excessive lordosis. There is relative clinical sparing of extraocular muscles and muscles of bladder and bowel sphincters as for unknown reasons these muscles lack dystrophin . By age 10–12 years, the child is typically unable to walk and confined to a wheel chair. By definition, boys who cease walking by age 13 years have DMD—whereas boys who are able to still walk independently to age 16 years have BMD. Deep tendon reflexes are lost, and joint contractors appear at the hip flexors and heel cords. By late teens, the weakness is profound, scoliosis is marked, and joint contractures are frequent. About 25 % of children have IQ scores below 75, and the average IQ score is one standard deviation below the mean, from 80 to 90. Cognitive involvement is not progressive over the course of the disease. Some children develop smooth muscle involvement with gastroparesis and constipation. Cardiomyopathy, cardiac muscle damage, slowly develops. Kyphoscoliosis and weakness of respiratory muscles produce a decreasing lung vital capacity and low maximal inspiratory and expiratory pressures.
The terminal stages of DMD are characterized by recurrent pulmonary infections and often congestive heart failure. The age of death ranges from 10 to 30 years with a mean of 18 years. Only 5 % live beyond 26 years.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


