Fig. 7.1
Diagram of spinal cord and vertebral bodies
Spinal cord dysfunction results from traumatic, inflammatory, demyelinating, ischemic, nutritional, malignant, and degenerative conditions. Diseases affecting the spinal cord usually present as one of three clinical scenarios—two of which involve spinal cord parenchyma and one involves spinal cord roots. The first scenario is degenerative with loss of specific spinal cord elements as seen in amyotrophic lateral sclerosis (ALS) and subacute combined degeneration (vitamin B12 deficiency). The second is from a lesion at one level of the spinal cord as seen in back or neck trauma, cervical myelopathy from central protruding intervertebral disk or acute transverse myelitis . The final scenario is from compression of exiting spinal cord nerve roots producing a radiculopathy (sensory and motor dysfunction of a single dermatome/myotome) due to focal lesions such as posterolateral prolapse of a vertebral disk or a neurofibroma compressing a spinal cord root.
Clinical signs depend on the level of the spinal cord damage and whether the damage involves part or all of the cord. Thus, to understand the clinical signs produced by lesions in spinal cord parenchyma, one must know the differences between upper motor neuron and lower motor neuron dysfunction (Fig. 7.2) and anatomic location and function of key spinal cord tracts (Fig. 7.3, and Tables 7.1, 7.2).
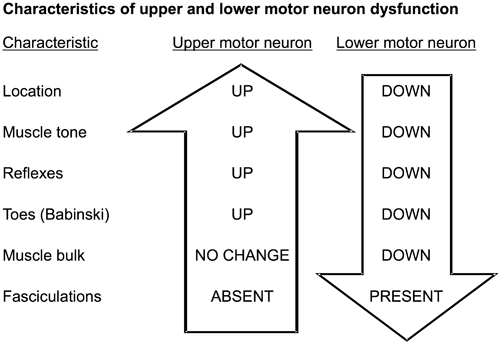
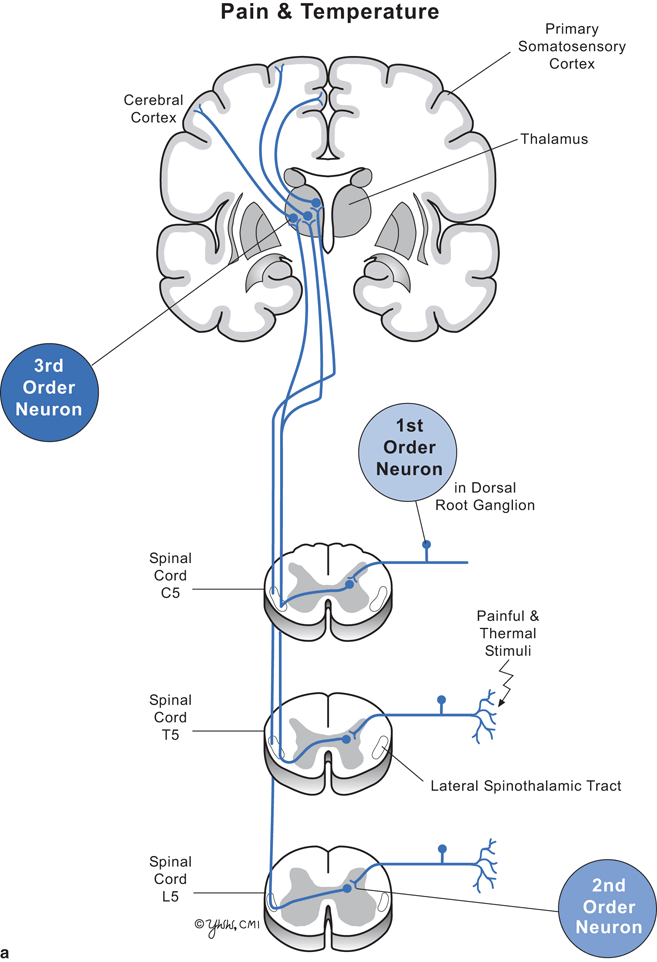
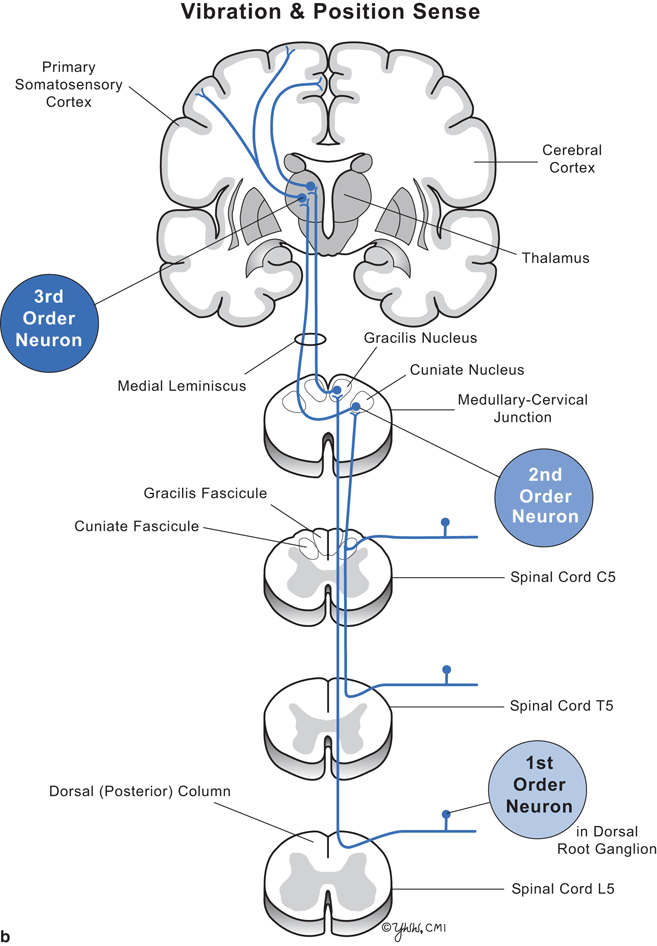
Fig. 7.2
Characteristics of upper and lower motor neuron disease
Fig. 7.3
Diagram of cervical, thoracic, and lumbar cord with pathways. a Pain and temperature. b Vibration and position sense
Table 7.1
Major spinal cord tracts and their function
Tract | Direction | Function | Location in spinal cord | Point of tract crossing to opposite side of spinal cord or medulla |
|---|---|---|---|---|
Corticospinal | From brain | Motor | Lateral | Medulla |
Spinothalamic | To brain | Pain, temperature | Lateral | Near site of spinal cord entry |
Dorsal column | To brain | Vibration and position sense | Posterior | Medulla |
Table 7.2
Major spinal cord neuronal groups
Name | Function | Location in spinal cord |
|---|---|---|
Anterior horn | Lower motor neurons | Ventrolateral gray matter |
Dorsal horn | Modulation of afferent sensory impulses | Dorsolateral gray matter |
Intermediolateral horn | Sympathetic neurons | Intermediolateral gray matter of thoracic spinal cord |
Lateral horn | Parasympathetic neurons | Lateral gray matter of sacral spinal cord |
Amyotrophic Lateral Sclerosis
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder that is uniformly fatal affecting primarily upper and lower motor neurons. Both the clinical and pathological features, as well as recent advances in genetics, suggest considerable heterogeneity in the phenotype of ALS. The disease is commonly known as Lou Gehrig’s disease, named for the famous baseball player who developed ALS in 1939. The term “amyotrophic” refers to muscle atrophy and “lateral sclerosis” refers to hardening noted on palpation of the spinal cord from gliosis following degeneration of the lateral corticospinal tracts. Not only is the cause of most cases unknown, but there is also controversy about where the disease begins. A dying-forward hypothesis argues the degeneration begins in motor neurons in the cortex and proceeds downward to involve the anterior horn cells (the motor neurons in the spinal cord). The dying-backward hypothesis proposes ALS begins at the muscle or neuromuscular junction with a loss or abnormal production of a trophic factor critical for the viability of motor neurons.
The incidence of ALS is 2 cases per 100,000 person years with a lifetime prevalence of 4 cases per 100,000. About 5,000 Americans are diagnosed with ALS annually. Analysis of incidence studies suggests the incidence is remaining stable. The peak age of onset ranges between 40 and 60 years with a marked decline after 75 years. The male-to-female ratio is 1.5:1. Most cases are sporadic but 5 % are hereditary.
In the typical case, the diagnosis is straightforward. For atypical onset cases, the differential diagnosis includes cervical spondylotic myeloradiculopathy, multifocal motor neuropathy, X-linked spinobulbar muscular atrophy (Kennedy’s disease), thyrotoxicosis, and elongated spinal cord tumors.
Pathophysiology
The pathogenesis of sporadic ALS is unknown. The main clinical and pathologic features are death of upper and lower motor neurons of the spinal cord and brainstem. Motor neurons may be more vulnerable to a variety of pathological processes because they: (1) are large, (2) have long large axons, (3) have high metabolic demands requiring optimal mitochondrial function, (4) are vulnerable to oxidative stresses and dysregulation of calcium homeostasis, and (5) are reliant on efficient synaptic glutamate reuptake transport mechanisms. No animals have been recognized to spontaneously develop a clinical or pathological picture of ALS. Thus, the several current hypotheses are derived from studies involving genetic causes for ALS, animal models of the genetic mutations, and sporadic human cases. The most common mutation (20 % of total cases) producing hereditary ALS stems from a missense mutation in the Cu/Zn superoxide dismutase (SOD1) gene. The mutated superoxide dismutase protein acts through an unknown “gain of function” producing the accumulation of superoxide with abnormal free radical formation in the spinal cord. Mitochondrial disruption is also thought to play a role as altered mitochondrial morphology is seen in skeletal muscle and spinal cord motor neurons in ALS. Excitotoxicity is thought to contribute to motor neuron injury mainly from excessive glutamate, the main excitatory transmitter in the CNS, possibly by an impaired uptake mechanism. Elevated CSF glutamate levels occur in some ALS patients, and riluzole, a drug that inhibits synaptic glutamate release, produces a modest prolongation of survival. Pathological protein aggregation occurring as ubiquitinated inclusions in the cytoplasm of neurons and glia is a cardinal feature of ALS. A major protein constituent of the inclusions is TAR DNA-binding protein 43 (TDP-43), which is important in RNA and DNA processing. Several ALS mutations are now recognized to affect this protein. Other hypotheses include dysregulated transcription and RNA processing, endoplasmic reticulum stress, impaired axonal transport, and neuroinflammation.
Motor neurons of the spinal cord and brainstem show simple atrophy and accumulation of intracytoplasmic ubiquitinated inclusion bodies containing TAR DNA-binding protein 43 leading to cell death and secondary astrocytic gliosis. There is a greater than 50 % reduction in the number of large motor neurons in the anterior horns of the cervical and lumbar spinal cord with a corresponding loss of large myelinated axons in the ventral roots and peripheral nerves going to the limbs. Interestingly, there is little anterior horn cell loss in the thoracic and sacral spinal cord, accounting for relative preservation of autonomic function and bladder and bowel function. The lower cranial nerves leading to bulbar muscles of the face (especially CN 7, 9, 10, 11, and 12) are more affected than cranial nerves supplying oculomotor muscles (CN 3, 4, and 6).
In the cerebral cortex, there is depletion of giant pyramidal neurons (Betz cells) and motor neurons in the fifth layer of the motor cortex with secondary degeneration of the corticospinal tracts.
Loss of lower motor neurons leads to muscle fiber denervation and weakness. Studies show that weakness progresses at a relatively constant rate throughout most of the disease. In early stages of the illness, a compensatory mechanism enables denervated muscles to become reinnervated and temporarily regain function. Following death of a motor neuron, a denervated muscle fiber produces an unknown trophic factor that signals adjacent motor axons to send a branch axon (sprouting) toward the denervated fiber with subsequent reinnervation of the fiber. This compensatory mechanism eventually fails when the replacement motor neuron dies.
There is now evidence that ALS produces more than damage just to the motor system. It has been recognized that some ALS patients share clinical and pathological features with frontotemporal lobe degeneration and three mutations have been linked to both conditions.
Major Clinical Features
Patients with ALS are typically divided into 4 phenotypes. Two-thirds of patients present with limb weakness, while 30 % present with bulbar dysfunction (dysarthria or dysphagia.) The remaining one-third develops respiratory dysfunction at disease onset, or “pure upper motor or lower motor neuron” signs.
Symptoms and signs of ALS are primarily those of a progressive upper and lower motor neuron loss. Loss of motor cortex neurons (upper motor neurons) leads to: (1) limb spasticity, (2) hyperactive reflexes, (3) Babinski signs, (4) limb paresis, and (5) pseudobulbar palsy (dysarthria, dysphagia and pseudobulbar affect with emotional reactions that are labile, exaggerated, and often inappropriate).
Loss of anterior horn neurons (lower motor neurons) causes the following: (1) arm and leg muscle weakness that is symmetrical or slightly asymmetrical; (2) muscle atrophy; (3) widespread muscle fasciculations; (4) eventual loss of reflexes; and (5) respiratory weakness from loss of phrenic nerve neurons to the diaphragm and neurons to accessory respiratory muscles.
Loss of bulbar lower motor neurons produces the following: (1) atrophy of the tongue (small tongue with serrated edges); (2) fasciculations of tongue; (3) atrophy of masseter muscle and muscles involved in swallowing producing dysphagia that can cause choking and malnutrition; (4) dysarthria making speech slow and difficult to understand; and (5) mild to moderate lower facial muscle weakness and atrophy.
Of note, muscles involved in eye movements and bladder and bowel function are seldom involved. Sensation and autonomic nerve function are preserved.
The weakness usually begins distally in the limbs and progresses to involve bulbar muscles, but in 20 % the process begins in bulbar muscles. Upper motor neuron signs may predominate early but subside as the lower motor neuron disease progresses and masks them.
Cognitive impairment identified by careful neuropsychological testing develops in about 25 % but is seldom noted by physicians as it is often subtle and coincides with the patient becoming depressed, fatigued from their weakness, or developing dyspnea. When recognized, the cognitive impairment is characterized by a personality change, irritability, poor insight, impulsivity, and impaired judgment. Frank dementia or aphasia is uncommon.
Typically because the onset of ALS is slow and variable, the correct diagnosis is often delayed for a year while other diagnoses are pursued.
Major Laboratory Findings
No specific diagnostic test for ALS exists. The hemogram, electrolytes, B12, and liver and renal function studies are normal. Serum creatine kinase may be mildly elevated, especially in rapidly progressive disease. CSF is usually normal but may have a slightly elevated protein level.
The EMG shows evidence of widespread denervation involving muscles of multiple myotomes. Common findings include (1) fibrillations and positive sharp waves, (2) reduced motor unit firing rates, and (3) neurogenic motor units of long duration, multiple phases, and increased amplitude (large polyphasic motor unit potentials). Early motor nerve conduction velocity is normal but slows later in the illness due to loss of the large myelinated axons that have the fastest conduction. However, evidence of slowing of conduction velocities from demyelination should not be present.
The diagnosis of ALS is usually established in a patient who has upper and lower motor neuron clinical signs involving arms and legs, and ideally, bulbar muscles plus EMG confirmation of lower motor neuron findings in several limbs. Usually a cervical MRI is done to rule out cervical spinal cord disease.
A muscle biopsy is occasionally done when the diagnosis is uncertain. Involved skeletal muscle fibers show changes typical for denervation that include pyknotic nuclear clumps involving sarcolemmal nuclei and atrophy of fibers leading to small angulated fibers with concave borders that are all the same fiber type. Early, the atrophic fibers are scattered but later they occur in clusters called “group atrophy”. The cluster has all the same fiber type staining. Normally, a cross section of skeletal muscle stained for fiber type presents a checkerboard appearance of type I and II fibers. Group atrophy is seen when muscle fibers lose their original motor unit, gain a new motor unit from sprouting of an adjacent motor nerve that in turn dies leaving a group of muscle fibers all of the same type.
Genetic testing can be done in patients with a family history of ALS-like syndromes. Advances in neuroimaging made possible the finding of subtle structural changes both within and outside the motor and premotor cortex, but these are not part of the diagnostic criteria currently.
Principles of Management and Prognosis
No drug has been found that stops the progressive loss of motor neurons. However, riluzole increases the survival of ALS patients by 3 to 6 months. Riluzole inhibits the release of the excitatory neurotransmitter glutamate . The goal of management is to make the patient as functional and comfortable as long as possible. Family and friends are valuable in supporting the patient and minimizing the reactive depression that commonly develops. Usually a multidisciplinary team approach is taken for the medical care. As patients weaken, crutches and wheelchairs are needed. When dysarthria becomes severe, assistive communication devices allow patients to continue expressing themselves. Managing dysphagia presents a challenge. Patients can swallow semisolid foods (e.g. pureed table foods) better than solid food or liquids. To prevent malnutrition and cachexia, a feeding gastrostomy or jejunostomy may be required. Inability to swallow leads to pooling of saliva in the posterior pharynx causing choking, drooling, and aspiration. Home suction equipment may be needed to minimize choking. Administration of anticholinergic drugs or botulinum toxin injections into the salivary glands may reduce production of saliva.
Respiratory weakness and failure become serious problems and usually trigger the terminal event of aspiration pneumonia. Early in the illness, a compassionate but frank interview with the patient and often family should focus on the patient’s terminal wishes and these should be placed in a living will. Non-invasive positive pressure ventilation improves dyspnea, increases the patient’s quality of life, and slightly extends the life expectancy. Few patients desire assisted invasive mechanical ventilation, such as via a tracheotomy, for the rest of their lives as they become progressively immobile. Respiratory measurements of forced vital capacity, the sniff nasal inspiratory pressure test, and overnight pulse oximetry are useful to follow a patient’s breathing capacity.
For sporadic ALS, the mean illness duration is 3 years but 10–15 % of patients live 5 years or more. Favorable prognostic indicators include age less than 50 years, lower limb onset, and long interlude from first symptom to diagnosis. Poorer prognostic indicators include bulbar onset, old age, malnutrition, known respiratory or cardiac disease, and impaired cognition.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


