Chapter 78 Disorders of Neuromuscular Transmission
Myasthenia Gravis
Epidemiology of Myasthenia Gravis
MG may begin at any age from infancy to very old age. Epidemiological studies report considerable variability in incidence and prevalence around the world (Meriggioli and Sanders, 2009). While methodological differences may explain some of this variability, biological and genetic factors may also play a role. Recent estimates indicate that the U.S. prevalence is approximately 20/100,000, 60,000 patients total (Phillips, 2004). Epidemiological studies have shown an increasing prevalence over the past 50 years, related to an increase in the frequency of diagnosis in elderly patients but also likely due to improved ascertainment, reduced mortality rates, and increased longevity of the population. Gender and age influence the incidence of MG; women are affected nearly three times more often than men before age 40, but the incidence is higher in males after age 50 and roughly equal during puberty. As the population ages, the average age at onset has increased correspondingly. More men than women are now affected, and the majority of MG patients in the United States are older than 50. Detailed population-based data on clinical and serological subtypes of MG (see Myasthenia Gravis Subtypes) are largely lacking.
Clinical Presentation of Myasthenia Gravis
Patients with MG seek medical attention for specific muscle weakness or dysfunction that typically worsens with activity and improves with rest. Although they may also have generalized fatigue or malaise, it is not usually the major or presenting complaint. Ptosis or diplopia is the initial symptom in approximately two-thirds of patients; nearly all will develop both within 2 years (Sanders and Massey, 2008). Difficulty chewing, swallowing, or talking is the initial symptom in one-sixth of patients, and limb weakness in 10%. Rarely, the initial weakness is limited to single muscle groups such as neck or finger extensors, hip flexors, or ankle dorsiflexors.
The course of disease is variable but usually progressive. Weakness remains restricted to the ocular muscles in approximately 10% to 15% of cases (see Ocular Myasthenia Gravis, later in this chapter), although a higher proportion has been reported in Asian populations (Meriggioli and Sanders, 2009). In the rest, weakness progresses to involve nonocular muscles during the first 3 years and ultimately involves facial, oropharyngeal, and limb muscles (generalized MG). Maximum weakness occurs during the first year in two-thirds of patients. Before the introduction of corticosteroids for treatment, approximately one-third of patients improved spontaneously, one-third became worse, and one-third died of the disease. Improvement, even remission, may occur early on but is rarely permanent (i.e., there is a subsequent relapse). Symptoms typically fluctuate over a relatively short period and then become more severe (active stage). Left untreated, an inactive stage follows the active stage, in which fluctuations in strength still occur but are attributable to fatigue, intercurrent illness, or other identifiable factors. After many years, untreated weakness becomes fixed, and the most severely involved muscles are frequently atrophic (burnt-out stage). Factors that worsen myasthenic symptoms are emotional upset, systemic illness (especially viral respiratory infections), hypothyroidism or hyperthyroidism, pregnancy, the menstrual cycle, drugs affecting NMT (see Treatment of Associated Diseases and Medications to Avoid, later in this chapter), and fever.
Physical Findings in Myasthenia Gravis
Ocular Muscles
 Most MG patients have weakness of ocular muscles (Box 78.1). (Videos of MG-related ocular phenomena [Videos 78.1 and 78.2] can be found at www.expertconsult.com.) Asymmetrical weakness of several muscles in both eyes is typical, the medial rectus being more frequently and usually more severely involved. The pattern of weakness is not localizable to lesions of one or more nerves, and the pupillary responses are normal. Ptosis is usually asymmetrical (Fig. 78.1) and varies during sustained activity. To compensate for ptosis, chronic contraction of the frontalis muscle produces a worried or surprised look. Unilateral frontalis contraction is a clue that the lid elevators are weak on that side (see Fig. 78.1). When mild, ocular weakness may not be obvious on routine examination and appear only upon provocative testing (i.e., sustained upward gaze). Eyelid closure is usually weak, even when strength is normal in all other facial muscles, and may be the only residual weakness in otherwise complete remission. This is usually asymptomatic unless it is severe enough to allow soap or water in the eyes during bathing. With moderate weakness of these muscles, the eyelashes are not “buried” during forced eye closure (Fig. 78.2). Fatigue in these muscles may result in slight involuntary opening of the eyes as the patient tries to keep the eyes closed; this is called the peek sign (see Fig. 78.2).
Most MG patients have weakness of ocular muscles (Box 78.1). (Videos of MG-related ocular phenomena [Videos 78.1 and 78.2] can be found at www.expertconsult.com.) Asymmetrical weakness of several muscles in both eyes is typical, the medial rectus being more frequently and usually more severely involved. The pattern of weakness is not localizable to lesions of one or more nerves, and the pupillary responses are normal. Ptosis is usually asymmetrical (Fig. 78.1) and varies during sustained activity. To compensate for ptosis, chronic contraction of the frontalis muscle produces a worried or surprised look. Unilateral frontalis contraction is a clue that the lid elevators are weak on that side (see Fig. 78.1). When mild, ocular weakness may not be obvious on routine examination and appear only upon provocative testing (i.e., sustained upward gaze). Eyelid closure is usually weak, even when strength is normal in all other facial muscles, and may be the only residual weakness in otherwise complete remission. This is usually asymptomatic unless it is severe enough to allow soap or water in the eyes during bathing. With moderate weakness of these muscles, the eyelashes are not “buried” during forced eye closure (Fig. 78.2). Fatigue in these muscles may result in slight involuntary opening of the eyes as the patient tries to keep the eyes closed; this is called the peek sign (see Fig. 78.2).
Box 78.1 Ocular Findings in Myasthenia Gravis
• Weakness usually involves one or more ocular muscles, without overt pupillary abnormality
• Weakness is typically variable, fluctuating, and fatigable
• Ptosis that shifts from one eye to the other is virtually pathognomonic of myasthenia gravis
• With limited ocular excursion, saccades are superfast, producing ocular “quiver”
• After downgaze, upgaze produces lid overshoot (“lid twitch”)
• Pseudo-internuclear ophthalmoplegia—limited adduction, with nystagmoid jerks in abducting eye
• In asymmetrical ptosis, covering the eye that has lid ptosis may relieve contraction of the opposite frontalis
•  Passively lifting a ptotic lid may cause the opposite lid to fall (“enhanced ptosis”) (see Video 78.2)
Passively lifting a ptotic lid may cause the opposite lid to fall (“enhanced ptosis”) (see Video 78.2)
• Edrophonium may improve only some of several weak ocular muscles; others may actually become weaker
• Edrophonium may relieve asymmetrical ptosis and produce retraction of the opposite lid from frontalis contraction
• The opposite lid may droop further as the more involved lid improves after edrophonium


Oropharyngeal Muscles
Myasthenic patients may have a characteristic facial appearance. At rest, the corners of the mouth often droop downward, giving a depressed appearance. Attempts to smile often produce contraction of the medial portion of the upper lip and a horizontal contraction of the corners of the mouth without the natural upward curling, which gives the appearance of a sneer (see Fig. 78.1).
Limb Muscles
Weakness begins in limb or axial muscles in about 20% of MG patients (Kuks and Oosterhuis, 2004). Any trunk or limb muscle may be weak, but some are more often affected than others. Neck flexors are usually weaker than neck extensors, and the deltoids, triceps, and extensors of the wrist and fingers and ankle dorsiflexors are frequently weaker than other limb muscles. Rarely, MG presents initially with focal weakness in single muscle groups, such as a “dropped head syndrome” due to severe neck extensor weakness or isolated vocal cord or respiratory muscle weakness. In untreated patients with long-standing disease, weakness may be fixed, and severely involved muscles may be atrophic, giving the appearance of a chronic myopathy; this is particularly likely in muscle-specific tyrosine kinase (MuSK) antibody–positive MG (see MuSK Antibody Myasthenia Gravis, later in this chapter).
Immunopathology of Myasthenia Gravis
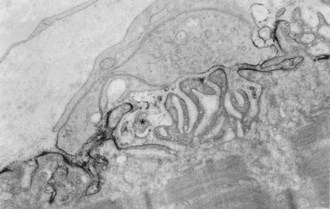
In about 80% to 85% of MG patients, weakness results from the effects of circulating anti-AChR antibodies. These antibodies bind to AChR on the terminal expansions of the junctional folds (Fig. 78.3) (Engel et al., 1977a) and cause complement-mediated destruction of the folds, accelerated internalization and degradation of AChR, and in some cases, they block ACh-AChR binding. Destruction of the junctional folds results in distortion and simplification of the postsynaptic region (see Fig. 78.4) and loss of functional AChR (Engel et al., 1977b). This leads to NMT failure and muscle weakness. MG is a paradigm for an antibody-mediated disease: the physiological abnormality is passively transferable by injection of MG immunoglobulin (Ig)G into mice, and clinical improvement follows removal of circulating antibodies by plasma exchange (see Treatment of Myasthenia Gravis, later in this chapter).
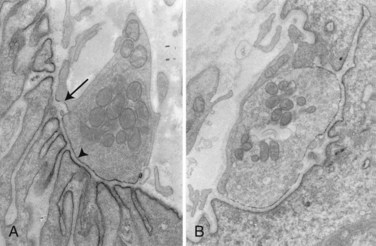
Patients with MG have increased numbers of CD4+ T cells, which regulate the production of AChR antibody (AChR-Ab). The α subunit of AChR contains the majority of T-cell recognition sites. These recognition sites may be different from those of the main immunogenic region that binding antibodies recognize. Sensitization to CD4+ T-cells spreads across the AChR complex as the disease progresses and most MG patients have T cells that recognize multiple epitopes on the AChR α-subunit (Conti-Fine et al., 1997). This epitope spread drives the synthesis of anti-AChR antibodies and accounts for the large and varied antibody repertoire of the myasthenic patient.
Recently, low-affinity IgG antibodies have been found in about two-thirds of MG patients who were seronegative using conventional anti-AChR and anti-MuSK antibody assays (Leite et al., 2008). These antibodies bind to AChRs that have been clustered into high-density arrays, suggesting that they have relatively low affinity and cannot bind strongly to AChR in solution but do bind to immobilized AChRs in a native conformation.
The Thymus in Myasthenia Gravis
The thymus is abnormal in most MG patients; 70% have lymphoid follicular hyperplasia, and more than 10% have a thymoma. Hyperplastic thymus glands from MG patients contain all the components necessary for the development of an immune response to the AChR: T cells, B cells, and plasma cells, as well as muscle-like myoid cells that express AChR. It is unlikely that the cellular alterations in the thymus are secondary to an ongoing peripheral immune response because they are absent in experimental autoimmune MG (Hohlfeld and Wekerle, 2008). In addition, thymocytes in culture spontaneously generate anti-AChR antibodies. These findings support the concept of an intrathymic pathogenesis and argue that the hyperplastic thymus is involved in the initiation of the anti-AChR immune response in patients with thymic hyperplasia. Thymic-derived AChR subunits may serve as an antigen for the autosensitization against the AChR.
Neoplastic epithelial cells in thymomas express numerous self-like antigens, including AChR, titin, and ryanodine receptor-like epitopes. MG-associated thymomas are also rich in autoreactive T cells. The regulation of potentially autoreactive T cells may be impaired in thymoma due to a deficiency in the expression of the autoimmune regulator gene (AIRE) and selective loss of T regulatory cells in human thymomas (Meriggioli and Sanders, 2009).
Myasthenia Gravis Subtypes
A number of MG subtypes (Table 78.1) may be identified based on the clinical presentation, age of onset, autoantibody profile, and thymic pathology (Meriggioli and Sanders, 2009). Interestingly, these subtypes appear to have unique genetic associations, strengthening the concept of distinct clinical entities and disease mechanisms.
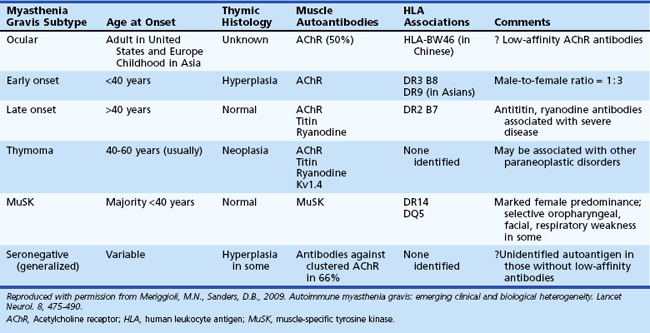
Ocular Myasthenia Gravis
Ptosis and/or diplopia are the initial symptoms of MG in up to 85% of patients (Grob et al., 2008), and almost all patients have both symptoms within 2 years of disease onset. Myasthenic weakness that remains limited to the ocular muscles is termed ocular myasthenia gravis (OMG) and accounts for approximately 10% to 15% of all MG in Caucasian populations. If weakness remains limited to the ocular muscles after 2 years, there is a 90% likelihood that the disease will not generalize. OMG is more common in Asian populations (up to 58% of all MG patients) (Zang et al., 2007).
Generalized Myasthenia Gravis
Patients with generalized myasthenia gravis (GMG) may have either early-onset (EOMG) or late-onset (LOMG) disease, with the cutoff age usually defined as age 40. EOMG patients are more often female and typically have anti-AChR antibodies and enlarged hyperplastic thymus glands. LOMG patients are more often male and may have antibodies to striated muscle proteins such as titin and the ryanodine receptor in addition to anti-AChR antibodies. The presence of these anti-muscle antibodies, particularly anti–ryanodine receptor antibodies, has been associated with more severe generalized or predominantly oropharyngeal weakness and frequent myasthenic crises (Romi et al., 2005). LOMG patients without thymoma usually have a normal or atrophic thymus, but relatively few histological studies are available in this age group, as thymectomy after age 50 is unusual.
MuSK-Antibody Myasthenia Gravis
Antibodies to MuSK have been reported in up to 50% of patients with GMG who lack AChR antibodies (Guptill and Sanders, 2010) and have recently been reported in OMG as well (Bau et al., 2006; Caress et al., 2005). The reported incidence of MuSK-antibody myasthenia gravis (MMG) varies among geographic regions, the highest being closer to the equator and the lowest closer to the poles (Vincent and Lang, 2006). Genetic or environmental factors (or both) presumably play a role in these differences. MMG predominantly affects females and begins from childhood through middle age. In some patients, the clinical findings are indistinguishable from anti-AChR-positive MG, with fluctuating ocular, bulbar, and limb weakness. However, many MMG patients have predominant weakness in cranial and bulbar muscles, frequently with marked atrophy of these muscles (Fig. 78.5). Others have prominent neck, shoulder, and respiratory weakness, with little or no involvement of ocular or bulbar muscles. Electrodiagnostic abnormalities may not be as widespread as in other forms of MG, and it may be necessary to examine different muscles to demonstrate abnormal NMT (Stickler et al., 2005). The potentially more limited distribution of physiological abnormalities also may limit the interpretation of microphysiological and histological studies in MMG, inasmuch as the muscles usually biopsied for these studies may be normal.
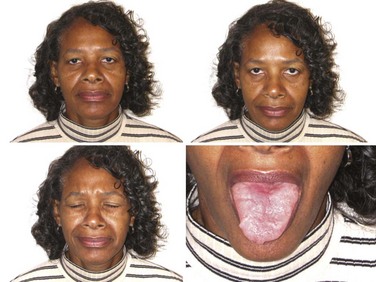
Many MMG patients do not improve with cholinesterase inhibitors (ChEIs); some actually become worse, and many have profuse fasciculations with these medications (Hatanaka et al., 2005). Disease severity tends to be worse, but most improve dramatically with PLEX or corticosteroids (Sanders et al., 2003). More immunosuppression is typically necessary, though long-term outcome is generally good (Guptill and Sanders, 2010). Thymic changes are absent or minimal (Lauriola et al., 2005; Leite et al., 2005), and the role of thymectomy in MMG is not yet clear (Guptill and Sanders, 2010; Sanders et al., 2003). The diagnosis of MMG may be elusive when the clinical features, electrodiagnostic findings, and response to ChEIs differ from typical MG.
Seronegative Myasthenia Gravis
MG patients who lack both anti-AChR and anti-MuSK antibodies (“double-seronegative MG”) are clinically heterogeneous. The true frequency of “seronegative MG” may be quite low, as certain patients may have low-affinity anti-AChR antibodies that can only be detected using specialized assays (see Immunopathology of Myasthenia Gravis, earlier).
Genetics of Myasthenia Gravis
Several correlations exist between MG and the human leukocyte antigen (HLA) genes. Certain HLA types (-DR2, -DR3, -B8, -DR1) predispose to MG (see Table 78.1), whereas others may offer resistance to disease. HLA-B8, -DR2, and -DR3 types occur more commonly in patients with EOMG; HLA-B7 and -DR2 in LOMG; and HLA-DR1 in OMG (see Table 78.1). MMG is associated with HLA-DR14-DQ5 (Niks et al., 2006). Different HLA associations have been reported in Asian MG patients, including an association of OMG with HLA-BW46 in Chinese patients (Meriggioli and Sanders, 2009). Non-HLA genes (PTPN22, FCGR2, CHRNA1) have also been found to be associated with MG; some are also associated with other autoimmune diseases and thus may represent a nonspecific susceptibility to autoimmunity. An exception to this is the CHRNA1 gene, which encodes the α subunit of the AChR and may provide pathogenetic clues specific for MG (Meriggioli and Sanders, 2009).
Diagnostic Procedures in Myasthenia Gravis
Edrophonium Chloride Test
The most important consideration in performance of the edrophonium test is the choice of endpoint. Only unequivocal improvement in strength of an affected muscle is acceptable as a positive result. For this reason, resolution of eyelid ptosis, improvement in strength of a single paretic extraocular muscle, or clear improvement of dysarthria have been proposed as the only truly valid endpoints because observed function in these muscles is largely independent of fluctuating effort (Fig. 78.6). Interpret changes in strength of other muscles cautiously, especially in a suggestible patient.
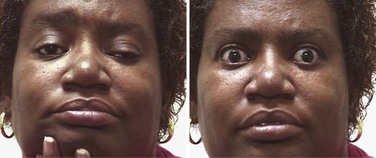
The edrophonium test is reported to be positive in 60% to 95% of patients with OMG and in 72% to 95% with GMG (Pascuzzi, 2003). Improvement after edrophonium is not unique to MG; it is also seen in congenital myasthenic syndromes, the Lambert-Eaton syndrome, intracranial aneurysms, brainstem lesions, cavernous sinus tumors, end-stage renal disease, and in muscle diseases affecting the ocular muscles.
 The optimal dose of edrophonium varies among patients and cannot be predetermined. In a study of OMG, the mean dose of edrophonium that gave a positive response was 3.3 mg for ptosis and 2.6 mg for ocular muscle dysfunction (Kupersmith et al., 2003). The lowest effective dose can be determined by injecting small incremental doses up to a maximum total of 10 mg. Inject an initial test dose of 2 mg, and monitor the response for 60 seconds. Subsequent injections of 3 and 5 mg may then be given, but if clear improvement is seen within 60 seconds after any dose, the test is positive, and no further injections are necessary (see Video 78.1). Weakness that develops or worsens after injection of 10 mg or less also indicates an NMT defect, as this dose will not weaken normal muscle.
The optimal dose of edrophonium varies among patients and cannot be predetermined. In a study of OMG, the mean dose of edrophonium that gave a positive response was 3.3 mg for ptosis and 2.6 mg for ocular muscle dysfunction (Kupersmith et al., 2003). The lowest effective dose can be determined by injecting small incremental doses up to a maximum total of 10 mg. Inject an initial test dose of 2 mg, and monitor the response for 60 seconds. Subsequent injections of 3 and 5 mg may then be given, but if clear improvement is seen within 60 seconds after any dose, the test is positive, and no further injections are necessary (see Video 78.1). Weakness that develops or worsens after injection of 10 mg or less also indicates an NMT defect, as this dose will not weaken normal muscle.
Common side effects of edrophonium are increased salivation and sweating, nausea, stomach cramps, and fasciculations. Serious complications (bradyarrhythmia or syncope) have been reported in only 0.16% of edrophonium tests (Ing et al., 2005). These symptoms generally resolve with rest in the supine position. Atropine (0.4-2 mg) should be available for intravenous (IV) injection if bradycardia is severe.
Autoantibodies in Myasthenia Gravis
Acetylcholine Receptor Antibodies
Assays measuring antibodies that react with AChR proteins are generally regarded as specific serological markers for MG. The most widely utilized test for MG is the AChR-Ab binding assay. This assay tests serum for binding to purified AChR from human skeletal muscle labeled with radioiodinated α-bungarotoxin. The reported sensitivity of this assay is approximately 85% for GMG and 50% for OMG (Stålberg et al., 2010). Nearly all thymomatous MG patients have elevated AChR antibodies.
AChR antibodies cross-link the AChRs in the membrane and increase their rate of degradation. The AChR-modulating antibody assay measures the rate of loss of labeled AChR from cultured human myotubes. About 10% of MG patients who do not have elevated binding antibodies have AChR-modulating antibodies. Many patients with thymomatous MG have high levels of AChR-modulating antibodies, often exceeding 90% loss of AChR (Vernino and Lennon, 2004).
Antistriational Muscle Antibodies
StrAbs are not pathogenic and are also found in one-third of patients with thymoma who do not have MG and in one-third of MG patients without thymoma. They are more frequent in older MG patients and in those with more severe disease, suggesting that disease severity is related to a more vigorous humoral response against multiple muscle antigens (Romi et al., 2005).
Electrodiagnostic Testing in Myasthenia Gravis
Repetitive nerve stimulation (RNS) is the most commonly used electrophysiological test of NMT. At low rates of stimulation (2-5 Hz), RNS depletes the store of readily releasable ACh at diseased motor end-plates, causing failure of NMT. Characteristically in MG, there is a decrementing response of at least 10% to trains of 2- to 3-Hz stimulation (see Chapter 32B). This may be present at baseline or after a period of exercise (postactivation exhaustion). Although a seemingly simple test, careful attention to proper technique is important to avoid technical errors. The sensitivity of RNS for diagnosing MG reportedly ranges from 53% to 100% in GMG and 10% to 48% in OMG (Meriggioli and Sanders, 2004; Stålberg et al., 2010). RNS is more likely to be abnormal in a proximal or facial muscle and in clinically weak muscles. For maximal diagnostic yield, test several muscles, particularly those that are weak. If RNS is normal and there exists a high suspicion for an NMJ disorder, perform SFEMG of at least one symptomatic muscle.
SFEMG (see Chapter 32B) is the most sensitive clinical test of NMT and shows increased jitter in some muscles in almost all patients with MG (Stålberg et al., 2010). Jitter is greatest in weak muscles but is usually abnormal even in muscles with normal strength. Sixty percent of patients with OMG show increased jitter in a limb muscle, but this does not predict the subsequent development of generalized myasthenia.
In the rare patient who has weakness restricted to a few limb muscles, only a weak muscle may show abnormal jitter. This is particularly true in some patients with MMG (Stickler et al., 2005) (see MuSK-Antibody Myasthenia Gravis, earlier).
Recently, measuring jitter with concentric needle electrodes (CNE) has been proposed as an alternative to the specially designed (reusable) single-fiber electrode (Stålberg and Sanders, 2009). Interpret the results with caution, particularly in borderline cases, as signals recorded with the CNE may represent the summation of more than one single-fiber action potential, which will decrease the apparent jitter.
Ocular Cooling
Myasthenic weakness typically improves with muscle cooling. This is the basis of the “ice-pack” test, in which cooling of a ptotic lid improves lid elevation. Assess improvement in ptosis after placing an ice pack over the ptotic eyelid, usually for 2 minutes. Positive responses can occur even when edrophonium tests are negative. A meta-analysis of six studies showed this test to have high sensitivity and specificity in MG, suggesting that it may be useful in patients with lid ptosis, particularly if the edrophonium test is negative or contraindicated (Larner, 2004).
Comparison of Diagnostic Techniques in Myasthenia Gravis
Plan diagnostic testing based on the clinical presentation and distribution of weakness (Table 78.2). The edrophonium test is often diagnostic in patients with ptosis or ophthalmoparesis but is less useful in assessing other muscles. The presence of serum AChR or anti-MuSK antibodies virtually ensures the diagnosis of MG, but their absence does not exclude it. RNS confirms impaired NMT but is frequently normal in mild or purely ocular disease. Almost all patients with MG have increased jitter, and normal jitter in a weak muscle excludes MG as the cause of the weakness. Neither electrodiagnostic test is specific for MG, because increased jitter, even abnormal RNS, occurs in other motor unit disorders that impair NMT.

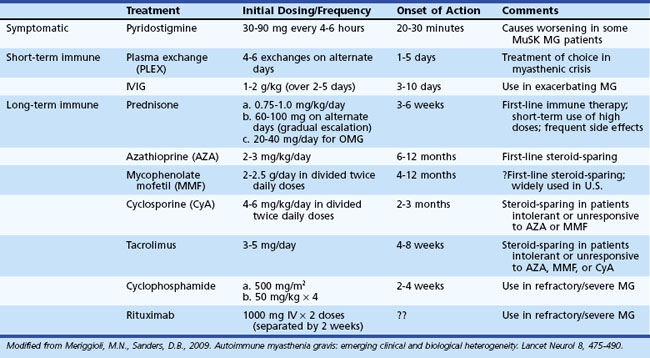
Treatment of Myasthenia Gravis
The outlook for patients with MG has improved considerably in recent years, largely due to advances in intensive care medicine and the use of immunomodulating agents. The therapeutic goal is to return the patient to normal function as rapidly as possible while minimizing the side effects of therapy. A number of therapeutic options are available (Table 78.3), but treatment must be individualized according to the extent (ocular versus generalized) and severity (mild to severe) of disease, and the presence or absence of concomitant disease (including but not limited to other autoimmune diseases and thymoma). The basis of treatment decisions for individual patients is the predicted course of disease and the predicted response to specific treatments. Successful treatment of MG requires close medical supervision and long-term follow-up. Consider the return of weakness after a period of improvement as a herald of further progression requiring reassessment of current treatment and evaluation for underlying systemic disease or thymoma.
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


