Focal (or multifocal) lesions of peripheral nervous system
Entrapment syndromes such as carpal tunnel syndrome
Ischemic neuropathy
Herpes zoster and postherpetic neuralgia
Trigeminal neuralgia
Vasculitis syndromes
Polyneuropathies
Diabetes mellitus
HIV neuropathy
Amyloidosis
B12 deficiency, beriberi
Hereditary sensory neuropathies
Toxic neuropathies
Toxins (arsenic, thallium, acrylamide, clioquinol, ethylene oxide)
Drugs (antiretrovirals, cisplatin, oxaliplatin, disulfiram, ethambutol, isoniazid, nitrofurantoin, thalidomide, vincristine, metronidazole)
Alcohol
Malignant (paraneoplastic syndromes, multiple myeloma)
Central nervous lesions
Spinal cord injury, syringomyelia
Brain infarction or malformation bleeds, esp. in thalamus and brainstem
Brain tumors, abscesses, subdural hematoma, meningitis
Complex mixed pain syndromes with both tissue damage and neuropathic pain
Disk protrusion producing low back or neck pain with radiculopathy
Cancer pain
Postsurgical operations
Regional pain syndromes such as complex regional pain syndrome, causalgia
Phantom limb pain or amputation stump pain
Headachea
Tension
Migraine
Uncommon (cluster, occipital neuralgia, temporal arteritis)
Normal Pain Pathways
Pain pathways appeared at different times in evolution. The most primitive system uses specific polymodal nociceptors that are activated by a specific type of high-intensity mechanical, chemical (such as chili, garlic, mustard, horseradish), or thermal stimulus. Thus, nociceptor nerves differ from other sensory nerves that respond to innocuous stimuli with a lower threshold to firing. The majority of nociceptive nerve fibers are small-diameter, unmyelinated C fibers that conduct slowly at 0.5–2.0 m/s The axon endings are free nerve terminals with no extracellular matrix capsule. These fibers fire continuously without decay if the noxious stimulus is maintained. Activation of nociceptive C fibers is appreciated as a burning, uncomfortable, and poorly localized pain.
The cell bodies of all C fibers are located in the dorsal root ganglia. A single axon emerges from each cell body, with the axon immediately bifurcating into two fibers, one projecting to the periphery innervating various body regions (e.g. the skin), while the other fiber projects to the central nervous system (spinal cord, or brainstem for the head and neck). The centrally projecting nerve terminals communicate to nociceptive-specific projection neurons in the superficial layers (lamina I and II) of the spinal cord dorsal horn of several rostro-caudal adjacent segments. Centrally projecting C-fibers additionally contact excitatory and inhibitory interneurons present in lamina II. Many inhibitory interneurons are spontaneously active, maintaining tonic inhibitory control over dorsal horn processing. C-fiber terminals release glutamate and substance P as their excitatory neurotransmitters . The incoming signal may be diminished or inhibited by interactions from endorphin-releasing interneurons (endogenous opioids), or influenced by inhibitory or facilitatory axons descending from supraspinal regions such as the somatosensory cortex, hypothalamus, midbrain periaqueductal gray, and pons .
Nociceptive spinal neurons that become activated by pain-related stimuli project to various sites in the brainstem and thalamus before reaching the cortex for the conscious perception of pain. The most important ascending pathways for pain processing consist of direct spinal projections to the thalamus, direct spinal projections to homeostatic control areas in the medulla, and projections to the hypothalamus and ventral forebrain.
The most recent evolutionarily pain pathway conducts nociceptive signals from the body to the brain in a similar manner to C-fibers following noxious mechanical, thermal, or chemical input that ultimately reach the cerebral cortex, resulting in conscious awareness of pain. These peripheral pain fibers are small-diameter, thinly myelinated Aδ fibers that conduct at 5–30 m/s. Thus, this pathway system is more rapid than the primitive C-fiber pathway and yields more precise localization of the pain source. Stimulation generates signals that are felt as sharp, pricking, localizable pain. The axons usually have dynamic firing rates that decline with time even if the stimulus is maintained (known as accommodation). As with C-fibers, Aδ fibers travel through the dorsal root ganglion , terminate in the superficial layers of the dorsal horn of the adjacent segments of spinal cord, and release excitatory neurotransmitters such as glutamate. Again, interneurons modulate further transmission of the pain signal. Second-order axons in the spinothalamic pain pathway cross the spinal cord midline and travel to the contralateral spinothalamic tract to terminate at the thalamus (ventral posterior lateral and central lateral nuclei). Third-order axons then travel to the somatosensory cortex, somatosensory, anterior cingulate, and insular cortices. How pain signals reach conscious perception is poorly understood.
Neuropathic Pain Mechanisms
The pathophysiologic knowledge of NP is expanding, but the steps that initiate the process are still poorly understood . Physiologic changes producing NP develop at several anatomic locations including the damaged nerves, dorsal root ganglia, spinal cord dorsal column root entry zone, brainstem, thalamus, and cerebral cortex. It appears that the anatomic location of the pathologic changes varies from person to person yet often produces similar symptoms and appears to respond similarly to the pain medications.
Peripheral sensitization begins with stimulus-evoked plasticity of the peripheral nociceptors initiated from inflammatory mediators released from injured nerves and inflammatory cells that sensitize the nociceptive terminal (Fig. 20.1). Cytokines, Substance P, bradykinin, serotonin, and prostaglandin sensitize injured and adjacent nociceptors to lower their threshold to fire—amplifying the intensity of the pain signal and expanding the skin area involved. The number of sodium channels increases in the damaged and adjacent intact sensory nerves, making the axon depolarize more easily. The net result of the peripheral nociceptive nerve plasticity is hypersensitivity and allodynia .
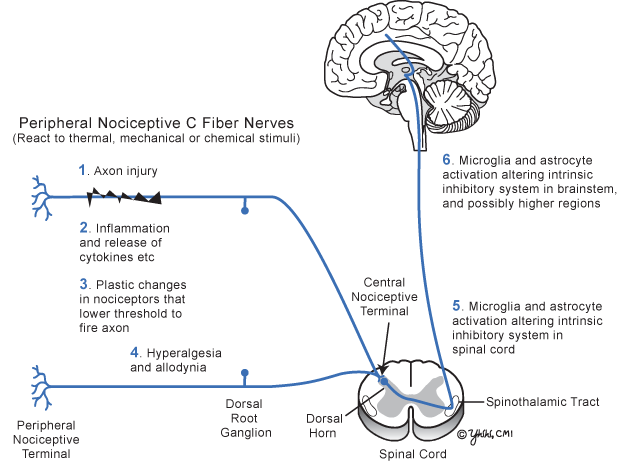
Fig 20.1
Current basic concepts of neuropathic pain development
At the spinal cord dorsal horn, brainstem, and thalamus, several complex events develop. Microglia, the resident macrophages of the central nervous system, become activated in the brainstem and possibly higher, releasing their cytokines, thus affecting the complex interaction of dorsal horn fibers. Adjacent astrocytes also become activated in the spinal cord and at the anterior cingulate cortex. Activated astrocytes surrounding the pain-related synapse affect pain transmission by inducing synaptic neuroplasticity that intensifies nociceptive transmission and eventually produces gliosis. The role of microglia and astrocytes in causing NP is growing in importance. In addition, negative feedback from inhibitory cortical fibers descending to the secondary dorsal horn pain neurons is diminished. Finally, changes in the central nervous system appear capable of inducing pain signals that did not originate in the peripheral nervous system. These are called central pain syndromes (Table 20.1) .
Predisposing factors for chronic pain development have been identified and include genetic factors, poor coping skills, depression, intensity of the original tissue damage, and female gender.
In summary (Fig. 20.1), NP usually begins following injury to peripheral nociceptive axons resulting in local inflammation that induces complex plastic changes in the peripheral nociceptor resulting in a lower threshold for the injured axon and adjacent axons to fire. At the spinal cord dorsal horn and brainstem, secondary changes develop from the activation of microglia and astrocytes that alter the normal negative feedback control of pain signals, which adds to aberrant pain signal transmission and often perpetuates pain, even when the peripheral nerve injury has recovered.
Neuropathic Pain Management
As the physiology of NP is becoming better understood, there is improved understanding of how the conventional pain medications may work (Table 20.2). Unfortunately, we have not yet found methods to prevent or terminate the entire NP process . The most commonly administered, evidence-based, first-line medications for chronic pain are tricyclic antidepressants (amitriptyline and nortriptyline), gabapentin, opioids, and lidocaine patches. Each medication has multiple side effects so the drug type and dosage must be individualized for each patient. In general, always begin at a low dose and slowly increase to effectiveness over 1–2 weeks to reduce the side effects. It is useful to warn the patient that takes several weeks before the medications show benefit.
Table 20.2
Medications for chronic pain and their proposed mechanism of action
Medication for chronic pain | Proposed mechanism of action |
|---|---|
Tricyclic antidepressants Amitriptyline Nortriptyline Desipramine | Inhibition of reuptake of serotonin and/or norepinephrine, blocking sodium channels, and anticholinergic |
Selective serotonin and norepinephrine reuptake inhibitors (SSNRI) Duloxetine Venlafaxine | Inhibition of both serotonin and norepinephrine reuptake |
Opioid agonists | µ-receptor agonism in spinal cord dorsal horn and brainstem |
Anticonvulsants Carbamazepine Topiramate Valproate | Blockade of sodium channels at multiple locations |
Topical lidocaine | Blockade of sodium channels in peripheral nerve axons |
It is frustrating that in spite of a wide variety of available pain medications, less that 50 % of chronic pain patients achieve satisfactory pain relief with any drug combination.
Headache
Overview
For most types of head pain, the first and second divisions of the trigeminal nerve bring noxious signals to the brain and to awareness. Only certain parts of the central nervous system are innervated by pain fibers, including proximal cerebral vessels (especially internal carotid, proximal anterior, and middle cerebral arteries), dural blood vessels, large veins, sinuses, dura at base of brain, bone, and several cranial nerves (5,7,9,10). In addition, the scalp, skull muscles , sinus mucosa, and teeth are innervated by pain fibers. The brain parenchyma, ependyma, choroid plexus, pia, and dura over the convexity of the brain, however, are insensitive to pain. Somatic pain usually localizes to the exact area of injury (i.e. scalp laceration), while the pain from headache is more diffuse and localizes only to large regions of the head.
There are over 250 causes of headache. In general, a simple classification divides headaches into primary and secondary. Primary headaches (like tension-type, migraine, and cluster) are those headaches where pain is the primary symptom and no structural damage occurs to the brain. Secondary headaches (from tumor, infection, subdural hematoma, etc.) develop from structural damage to the skull or central nervous system masses causing secondary meningeal origin pain. Secondary headaches often produce other neurologic signs and symptoms.
The evaluation of a patient with headache usually requires a careful history with attention to its characteristics (Table 20.3). The exam should be thorough with attention for the presence of papilledema, neck stiffness, cranial nerve signs (especially the trigeminal nerve), or signs of sinus or tooth/mouth infection. For the headache to be considered primary, the neurologic exam should be normal. If the neurologic exam is abnormal, the headache may be secondary and the result of structural damage of the face, skull, meninges, or brain. If a secondary etiology of the headache is suspected and of recent origin, neuroimaging should be considered.
Table 20.3




Important questions in headache evaluation
Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
