Chapter 76 Disorders of Peripheral Nerves
Clinical Approach to Disorders of Peripheral Nerves
Pathological Processes Involving Peripheral Nerves
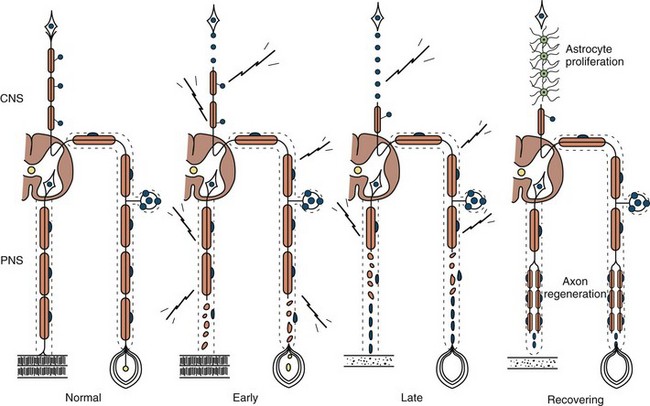
Any type of mechanical injury that causes interruption of axons leads to wallerian degeneration—that is, distal degeneration of axons and their myelin sheaths. Immediately following injury, motor weakness and sensory loss occur in the distribution of the damaged nerve, as well as complete loss of voluntary activity (with a complete lesion) or a decrease in motor unit action potential (MUAP) recruitment (with a partial lesion) on needle electromyogram (EMG). However, the axons remain excitable distally, since distal conduction failure is not completed until 10 to 11 days later as the distal nerve trunk becomes progressively unexcitable. On nerve conduction studies, the amplitude of the compound muscle action potential (CMAP) begins to decline by the second day after injury and reaches its nadir by the fifth to sixth. For sensory axons, the loss of sensory nerve action potential (SNAP) is delayed by another 2 to 3 days; distal SNAP remains normal for 5 to 6 days and then decreases rapidly to reach its nadir by 10 to 11 days after injury (see Fig. 32B.9 in Chapter 32B). The temporal sequence of wallerian degeneration is length dependent, occurring earlier in shorter than in longer distal nerve stumps. Denervation potentials (fibrillation potentials) are typically seen on needle EMG in some affected muscles 10 to 14 days after injury and become full after 3 weeks from acute nerve injury. Axonal interruption initiates secondary morphological changes of the nerve cell body, termed chromatolysis, and the proximal axonal caliber becomes smaller. Regeneration from the proximal stump begins as early as 24 hours following transection but proceeds slowly at a maximal rate of 2 to 3 mm/day and is often incomplete. The quality of recovery depends on the degree of preservation of the Schwann cell/basal lamina tube and the nerve sheath and surrounding tissue, the distance of the site of injury from the cell body, and the patient’s age.
Axonal degeneration (or axonopathy), the most common pathological reaction of peripheral nerve, signifies distal axonal breakdown and is presumably caused by metabolic derangement within neurons or ischemia. Systemic metabolic disorders, toxin exposure, vasculitis, and some inherited neuropathies are the usual causes of axonal degeneration. The myelin sheath breaks down concomitantly with the axon in a process that starts at the most distal part of the nerve fiber and progresses toward the nerve cell body, hence the term dying-back or length-dependent polyneuropathy (Fig. 76.1). A similar sequence of events may occur simultaneously in centrally directed sensory axons, resulting in distal degeneration of rostral dorsal column fibers. The selective length-dependent vulnerability of distal axons could result from failure of the perikaryon to synthesize enzymes or structural proteins, from alterations in axonal transport, or from regional disturbances of energy metabolism. In some axonopathies, alterations in axon caliber, either axonal atrophy or axonal swelling, may precede distal axonal degeneration. Clinically, dying-back polyneuropathy presents with symmetrical distal loss of sensory and motor function in the lower extremities that extends proximally in a graded manner. The result is sensory loss in a stocking-like pattern, distal muscle weakness and atrophy, and loss of distal limb myotatic reflexes. Axonopathies result in low-amplitude SNAPs and CMAPs, but they affect distal latencies and conduction velocities only slightly. Needle EMG of distal muscles shows acute and/or chronic of denervation changes (see Chapter 32B). Because axonal regeneration proceeds at a maximal rate of 2 to 3 mm/day, recovery may be delayed and is often incomplete.
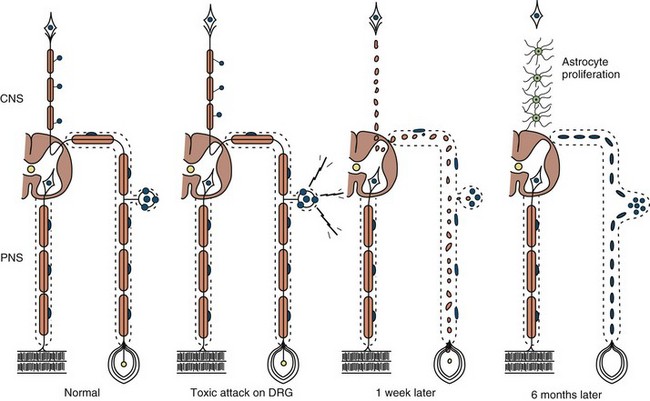
Neuronopathy designates loss of nerve cell bodies with resultant degeneration of their entire peripheral and central axons. Either anterior horn or dorsal root ganglion cells may be affected. When anterior horn cells are affected, as in anterior poliomyelitis or motor neuron disease, focal weakness without sensory loss results. Sensory neuronopathy, or dorsal polyganglionopathy, means damage to dorsal root ganglion neurons that results in sensory ataxia, sensory loss, and diffuse areflexia (Fig. 76.2). A number of toxins such as organic mercury compounds, doxorubicin, and high-dose pyridoxine produce primary sensory neuronal degeneration. Immune-mediated inflammatory damage of dorsal root ganglion neurons occurs in paraneoplastic sensory neuronopathy and Sjögren syndrome. It is often difficult to distinguish between neuronopathies and axonopathies on clinical grounds alone. Once the pathological processes are no longer active, sensory deficits become fixed, and little or no recovery takes place.
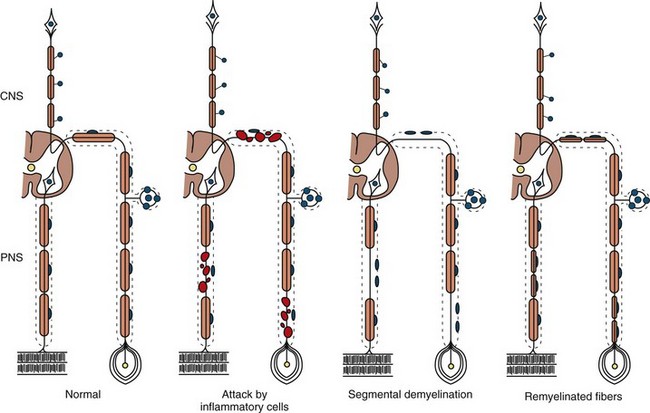
The term segmental demyelination (or myelinopathy) implies injury of either myelin sheaths or Schwann cells, resulting in breakdown of myelin with sparing of axons (Fig. 76.3). This occurs mechanically by acute nerve compression or chronic nerve entrapment and in immune-mediated demyelinating neuropathies and hereditary disorders of Schwann cell/myelin metabolism. Primary myelin damage may be produced experimentally by myelinotoxic agents such as diphtheria toxin or by acute nerve compression. Remyelination of demyelinated segments usually occurs within weeks. The newly formed remyelinated segments have thinner-than-normal myelin sheaths and internodes of shortened length. Repeated episodes of demyelination and remyelination produce proliferation of multiple layers of Schwann cells around the axon, termed an onion bulb. The physiological consequence of acquired demyelination, such as in inflammatory or compressive demyelination but not hereditary myelinopathies, is conduction block, which results in loss of the ability of the nerve action potential to reach the muscle, thereby producing weakness. Because the axon remains intact, there is little muscle atrophy. Relative sparing of temperature and pinprick sensation in many demyelinating polyneuropathies reflects preserved function of unmyelinated and small-diameter myelinated fibers. Early generalized loss of reflexes, disproportionately mild muscle atrophy in the presence of proximal and distal weakness, neuropathic tremor, and palpably enlarged nerves are all clinical clues that suggest demyelinating polyneuropathy. Nerve conduction studies or analysis of single teased nerve fiber preparations stained with osmium can confirm demyelination. Demyelination is present if motor and sensory nerve conduction velocities (NCVs) are reduced to less than 70% of the lower limits of normal, with relative preservation of response amplitudes. The presence of partial motor conduction block, temporal dispersion of CMAPs, and marked prolongation of distal motor and F-wave latencies are all features consistent with acquired demyelination (see Chapter 32B). Recovery depends on the extent of remyelination, and therefore clinical improvement may occur within days to weeks. It should be noted that in many demyelinating neuropathies, axonal degeneration may also coexist.
Classification of Peripheral Nerve Disorders
There are several patterns of peripheral nerve disease (Box 76.1). Brachial, lumbar, and sacral plexopathy are discussed in Chapter 75, and radiculopathies are discussed in Chapter 74.
Box 76.1 Classification of Peripheral Nerve Disease
Diagnostic Clues from the History
Symptoms of autonomic dysfunction can be helpful in directing attention toward specific neuropathies that have prominent autonomic symptoms. It is important to ask about orthostatic intolerance (lightheadedness, presyncopal symptoms or syncope), reduced or excessive sweating, heat intolerance, and bladder, bowel, and sexual dysfunctions. Anorexia, early satiety, nausea, and vomiting are symptoms suggestive of gastroparesis. The degree of autonomic involvement can be documented by noninvasive autonomic function studies (see Chapter 77).
Diagnostic Clues from the Examination
In multiple mononeuropathies (mononeuropathy multiplex), the neurological findings should point to simultaneous or sequential damage to two or more noncontiguous peripheral nerves. Confluent multiple mononeuropathies, such as with involvement of the fibular and tibial nerves or median and ulnar nerves, may give rise to motor weakness with sensory loss that can simulate a length-dependent peripheral polyneuropathy. EDX studies ascertain whether the primary pathological process is axonal degeneration or segmental demyelination (Box 76.2). Approximately two-thirds of patients with multiple mononeuropathies display a picture of axonal damage. Ischemia caused by systemic or nonsystemic vasculitis or microangiopathy in diabetes mellitus should be considered. Other less common causes are disorders affecting interstitial structures of nerve, namely infectious, granulomatous, leukemic, or neoplastic infiltration, including Hansen disease (leprosy) and sarcoidosis. In the event focal demyelination or motor conduction block leads to multiple mononeuropathies, multifocal acquired demyelinating sensory and motor neuropathy (Lewis-Sumner syndrome), multifocal motor neuropathy, or HNPP should be considered.
Box 76.2 Causes of Multiple Mononeuropathies
Motor deficits tend to dominate the clinical picture in acute and chronic inflammatory demyelinating polyneuropathies, hereditary motor and sensory neuropathies, and in neuropathies associated with osteosclerotic myeloma, porphyria, lead toxicity, organophosphate intoxication, and hypoglycemia (Box 76.3). The distribution of weakness provides important information. Asymmetrical weakness without sensory loss suggests a motor neuronopathy such as motor neuron disease or multifocal motor neuropathy. The facial nerve can be affected in several peripheral nerve disorders (Box 76.4). In most polyneuropathies, the legs are more severely affected than the arms, with several notable exceptions (Box 76.5). Polyradiculoneuropathies cause both proximal and distal muscle weakness. For example, proximal and distal weakness is encountered in acute and chronic inflammatory demyelinating polyradiculoneuropathies, osteosclerotic myeloma, porphyria, and diabetic lumbar radiculoplexopathy. Nerve root involvement is confirmed by denervation in paraspinal muscles on needle EMG.
Box 76.3 Polyneuropathies with Predominantly Motor Manifestations
Acute motor axonal neuropathy*
Chronic inflammatory demyelinating polyradiculoneuropathy
Neuropathy with osteosclerotic myeloma
Diabetic lumbar radiculoplexopathy
Hereditary motor sensory neuropathies (Charcot-Marie-Tooth disease)
Box 76.5 Polyneuropathies with Predominantly Upper-Limb Motor Involvement
Autonomic dysfunction of clinical importance is seen in association with specific acute (e.g., GBS) or chronic (e.g., amyloid and diabetic) sensorimotor polyneuropathies. Rarely, an autonomic neuropathy can be the exclusive manifestation of a peripheral nerve disorder, without somatic nerve involvement (Box 76.6).
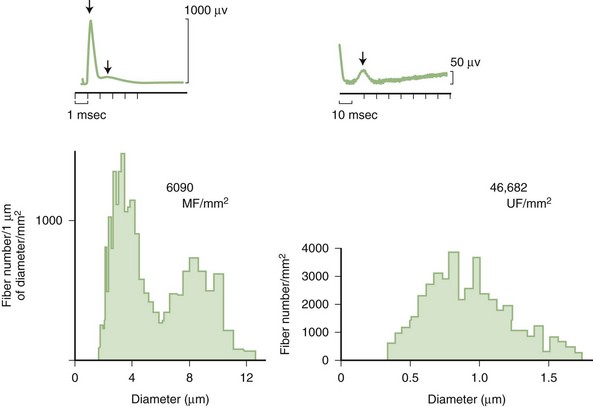
Predominant sensory involvement may be a feature of polyneuropathies caused by diabetes, carcinoma, Sjögren syndrome, dysproteinemia, acquired immunodeficiency syndrome (AIDS), vitamin B12 deficiency, celiac disease, inherited and idiopathic sensory neuropathies, and intoxications with cisplatin, thalidomide, or pyridoxine. Loss of sensation in peripheral neuropathies often involves all sensory modalities. However, the impairment may be restricted to selective sensory modalities in many situations, which makes it possible to correlate the type of sensory loss with the diameter size of affected afferent fibers (Fig. 76.4). Pain and temperature sensation are mediated by unmyelinated and small myelinated Aδ fibers, whereas vibratory sense, proprioception, and the afferent limb of the tendon reflex are subserved by large myelinated Aα and Aβ fibers. Light touch is mediated by both large and small myelinated fibers. In polyneuropathies preferentially affecting small fibers, diminished pain and temperature sensation predominate, along with spontaneous burning pain, painful dysesthesias, and autonomic dysfunction. There is preservation of tendon reflexes, balance, and motor strength, and hence few abnormal objective neurological signs are found on examination. A pattern of sensory loss that is very characteristic is distal loss of pinprick sensation, above which is a band of hyperalgesia (exaggerated pain from noxious stimuli), with normal sensation above this level. Relatively few disorders cause selective small-fiber neuropathies (Mendell and Sahenk, 2003) (Box 76.7). Selective large-fiber sensory loss is characterized by areflexia, sensory ataxia, and loss of joint position and vibration sense. Loss of joint position may also manifest as pseudoathetosis (involuntary sinuous movements of fingers and hands when the arms are outstretched and the eyes are closed) and/or a Romberg sign (disproportionate loss of balance with eyes closed compared with eyes open). Striking sensory ataxia, together with pseudoathetosis or asymmetrical truncal or facial sensory loss, directs attention to a primary disorder of sensory neurons or polyganglionopathies. The differential diagnosis of ataxic sensory neuropathies is limited (Box 76.8).
Box 76.8 Sensory Ataxic Neuropathies
Certain telltale signs of the skin and its appendages may direct the experienced examiner to a specific diagnosis (Table 76.1): alopecia is seen in thallium poisoning, tightly curled hair in giant axonal neuropathy; white transverse nail bands termed Mees lines in arsenic or thallium intoxications; purpuric skin eruptions of the legs in cryoglobulinemia and some vasculitides; skin hyperpigmentation or hypertrichosis in POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes); telangiectasias over the abdomen and buttocks in Fabry disease; enlarged yellow-orange tonsils in Tangier disease; pes cavus and hammer toes in CMT disease; and overriding toes and ichthyosis in Refsum disease.
Table 76.1 Neuropathies with Skin, Nail, or Hair Manifestations
| Disease | Skin, Nail, or Hair Manifestations |
|---|---|
| Vasculitis | Purpura, livedo reticularis |
| Cryoglobulinemia | Purpura |
| Fabry disease | Angiokeratomas |
| Leprosy | Skin hypopigmentation |
| Osteosclerotic myeloma (POEMS syndrome) | Skin hyperpigmentation |
| Variegate porphyria | Bullous lesions |
| Refsum disease | Ichthyosis |
| Arsenic or thallium intoxication | Mees lines |
| Thallium poisoning | Alopecia |
| Giant axonal neuropathy | Curled hair |
POEMS, Polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes.
Electrodiagnostic Studies
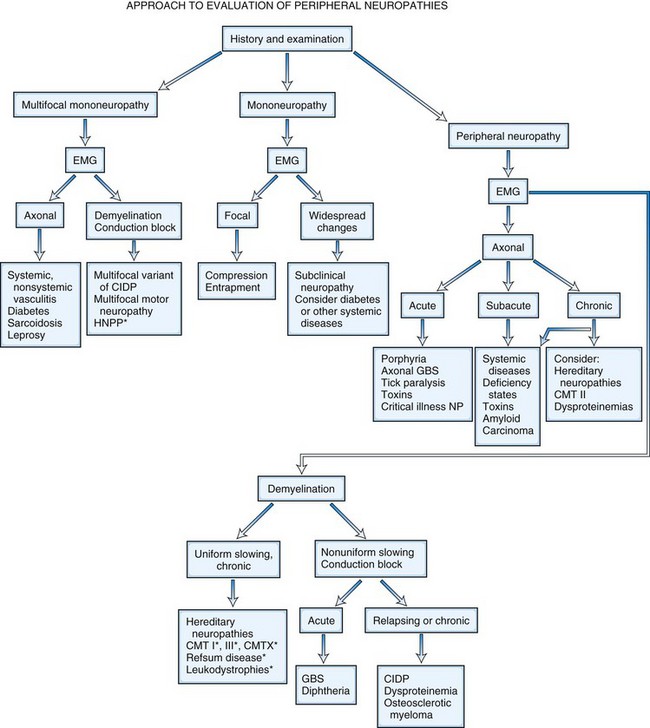
It is helpful to follow a decision-making pathway based initially on the overall pattern of distribution of deficits, followed by the electrophysiological findings, and finally the clinical course (Fig. 76.5). EDX studies, carefully performed and directed to the particular clinical situation, play a key role in the evaluation by (1) confirming the presence of neuropathy, (2) precisely locating focal nerve lesions, and (3) giving information as to the nature of the underlying nerve pathology (Gooch and Weimer, 2007; Wilbourn, A.J., 2002) (see Chapter 32B).
Because routine sensory nerve conduction studies assess only large myelinated fibers, such studies may be entirely normal in selective small fiber neuropathies. Quantitative sensory testing assessing cold and heat-pain thresholds, tests of sudomotor function, and skin biopsy with analysis of intraepidermal nerve fiber density may be helpful in confirming the unmyelinated nerve fiber abnormalities (Devigili et al., 2008). Since sweating mediated by unmyelinated sympathetic cholinergic fibers is often impaired, the quantitative sudomotor axon reflex (QSART) that evaluates sweating is a highly specific and sensitive method (sensitivity of 80%) to confirm small nerve fiber damage. Quantitative sensory testing assessing both vibratory and thermal detection thresholds has become a useful addition to the bedside sensory examination in controlled clinical trials. Its use in routine clinical practice remains limited because the test is still subjective in that it requires patient cooperation and is time consuming.
Nerve and Skin Biopsy
Skin punch or blister biopsies that demonstrate loss of intraepidermal nerve fibers is an alternative method for documenting small fiber neuropathy (Panoutsopoulou, 2009). Only unmyelinated intraepidermal networks of nerve fibers can be demonstrated by immunostaining with the panaxonal marker protein gene product 9.5, studied best with the use of confocal microscopy. Age, gender, and site of skin biopsy have a profound effect on epidermal nerve fiber density. The density of intraepidermal nerve fibers is reduced in skin biopsies obtained from patients with idiopathic, HIV-associated, diabetic, and other sensory neuropathies (Kennedy, 2004). Skin punch biopsy is most useful in patients with suspected small fiber neuropathy, when nerve conduction studies, are normal. The diagnosis of small-fiber neuropathy is best accomplished when at least two abnormal results are met, including positive clinical findings, quantitative sensory testing, QSART, and skin biopsy examinations (Devigili et al., 2008). Skin punch biopsy only detects the presence of skin nerve abnormalities and rarely leads to a specific etiological diagnosis. The skin biopsy also does not permit the study of myelinated fibers unless a thicker biopsy including dermis is obtained. Finally, unlike sural nerve biopsy, the interstitial pathological processes of the nerves cannot be studied.
Nerve biopsy (other than for the diagnosis of vasculitis and neoplasia) should be performed only in centers with established experience with the surgical procedure, handling of nerve specimens, and pathological technique; otherwise little useful information is likely to be obtained. The sural nerve is selected most commonly for biopsy, because the resultant sensory deficit is restricted to a small area over the heel and dorsolateral aspect of the foot, and because its morphology has been well characterized in health and disease. The superficial fibular nerve is an alternative lower-extremity cutaneous nerve suitable for biopsy and has the advantage of allowing simultaneous biopsy of the peroneus brevis muscle through the same incision. This combined distal nerve and muscle biopsy procedure increases the yield of identifying suspected vasculitis (Collins et al., 2000; Vital et al., 2006). In contrast, adding a proximal muscle (e.g., quadriceps) to a cutaneous nerve biopsy (e.g., sural) does not significantly increase the diagnostic yield compared to nerve biopsy alone (Bennett et al., 2008). In patients with proximal involvement of the lower limbs, the intermediate cutaneous nerve of the thigh combined with a muscle biopsy can be performed. When the risk of complication is increased from a biopsy of the lower limbs (e.g., in significant distal leg ischemia, edema) or the neuropathy is preferentially more pronounced in the upper limbs, a cutaneous nerve biopsy of superficial radial or antebrachial nerves may be performed. When the imaging studies indicate a plexus or nerve root pathological process (e.g., inflammatory, infiltrative), a fascicular biopsy of the affected nerve by an expert surgeon may provide invaluable information. Nerve biopsy has proved to be particularly informative when techniques such as single teased fiber preparations, semithin sections, ultrastructural studies, and morphometry are applied to quantitate the nerve fiber pathology. Nowadays, relatively few disorders remain in which a nerve biopsy is essential for diagnosis (Pleasure, 2007; Said, 2002) (Box 76.9). In general, nerve biopsy is most frequently diagnostic in suspected vasculitis, amyloid neuropathy, and leprosy. It is helpful in the recognition of CIDP, inherited disorders of myelin, and some rare axonopathies in which distinctive axonal changes occur, such as in giant axonal neuropathy and polyglucosan body disease. The availability of molecular genetic tests for several CMT neuropathies, HNPP, and familial transthyretin amyloidosis has decreased the necessity for nerve biopsy in these conditions.
Box 76.9 Indications for Nerve Biopsy
Nerve biopsy is an invasive procedure and is associated with as high as 15% complication rate—particularly minor wound infections, wound dehiscence, and stump neuromas. Approximately one-third of patients (particularly those without much sensory loss initially) report unpleasant sensory symptoms at the sural nerve biopsy site that are still present 1 year after the biopsy (Gabriel et al., 2000). The area of the original sensory deficit declines by 90% after 18 months because of collateral reinnervation (Theriault et al., 1998). The complications may be greater if substantial foot ischemia is present or if the patient smokes cigarettes.
Other Laboratory Tests
Several serum autoantibodies with reactivity to various components of peripheral nerve have been associated with peripheral neuropathy syndromes, and reference laboratories offer panels of nerve antibodies for sensory, sensorimotor, and motor neuropathies. It must be emphasized that the clinical relevance of most autoantibodies has not been established for patient treatment, and their use is not cost-effective (Vernino and Wolfe, 2007). Those of greatest clinical utility are listed in Table 76.2 (Kissel, 1998). An ever-increasing number of molecular genetic tests for inherited neuropathies is available at reference laboratories (see Hereditary Neuropathies, later).
Table 76.2 Neuropathies Associated with Serum Autoantibodies
| Autoantibody | Disease (% Positive) |
|---|---|
| ANTIBODIES AGAINST GANGLIOSIDES | |
| GM1 (polyclonal IgM) | Multifocal motor neuropathy (70%) |
| GM1, GD1a (polyclonal IgG) | Guillain-Barré syndrome (30%) |
| GQ1b (polyclonal IgG) | Miller Fisher variant of Guillain-Barré syndrome (>95%) |
| ANTIBODIES AGAINST GLYCOPROTEINS | |
| Myelin-associated glycoprotein (MAG; monoclonal IgM) | IgM monoclonal gammopathy of undetermined significance neuropathy (50%) |
| ANTIBODIES AGAINST RNA-BINDING PROTEINS | |
| Anti-Hu, antineuronal nuclear antibody 1 (ANNA1) | Malignant inflammatory polyganglionopathy (>95%) |
Ig, Immunoglobulin.
Mononeuropathies
Definition and Classification of Mononeuropathies
Peripheral nerve injuries are classified based on functional status of the nerve and histological findings. Seddon divided peripheral nerve injury into three classes: neurapraxia, axonotmesis, and neurotmesis. This classification remains popular, particularly among surgeons, because of its correlation to outcome (Seddon, 1975). Later, Sunderland (1991) revised the classification into five degrees that have better prognostic implications.
Neurotmesis (Fifth-Degree Nerve Injury)
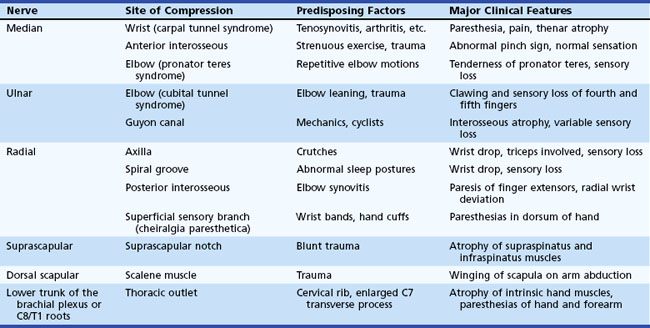
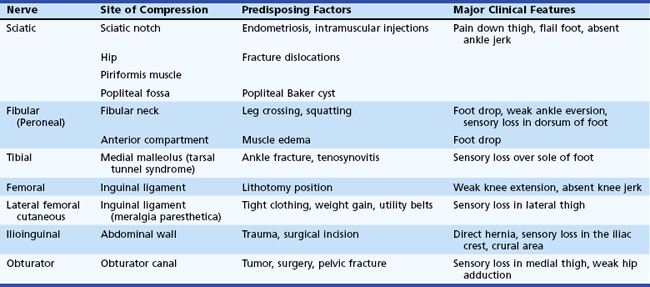
Entrapment neuropathy is defined as a mononeuropathy caused by focal compression or mechanical distortion of a nerve within a fibrous or fibro-osseous tunnel or less commonly by other structures such as bone, ligament, other connective tissues, blood vessels, or mass lesions. Compression, constriction, angulation, and stretching are important mechanisms that produce nerve injury at certain vulnerable anatomical sites (Tables 76.3 and 76.4). The term entrapment is a useful one in that it implies that compression occurs at particular sites where surgical intervention is often required to release the entrapped nerve, such as in the case of the median nerve at the wrist in moderate to severe carpal tunnel syndrome. Overuse has been implicated as the cause of entrapment neuropathies in certain occupations, including the playing of musical instruments by professional musicians.
The characteristic electrophysiological feature of entrapment neuropathy is either short-segment conduction delay (i.e., focal slowing) or conduction block across the site of entrapment (see Chapter 32B). In severe cases, wallerian degeneration gives rise to denervation and reinnervation in affected muscles. Nerve conduction studies together with needle EMG are essential for diagnosis and reliable documentation of the site and severity of nerve entrapment. Although plain radiography, computed tomography (CT), and magnetic resonance imaging (MRI) may be of occasional value in identifying rare structural abnormalities, these imaging procedures are not necessary for routine diagnosis.
Mononeuropathies of the Upper Extremities
Entrapment neuropathies of the upper extremities are shown in Table 76.3.
Median Nerve
Median Nerve Entrapment at the Wrist (Carpal Tunnel Syndrome)
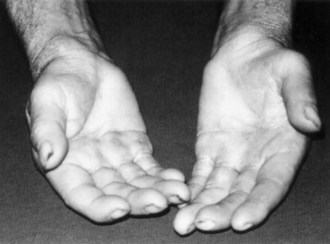
Symptoms consist of nocturnal pain and paresthesias, most often confined to the thumb, index, and middle fingers, but may be reported to involve the entire hand. Patients complain of tingling numbness and burning sensations, often awakening them from sleep. Referred pain may radiate to the forearm and even as high as the shoulder (Stevens et al., 1999). Symptoms are often provoked after excessive use of the hand or wrist or during ordinary activities such as driving or holding a phone, book, or newspaper, in which the wrist is assumed in either a flexed or extended posture. Objective sensory changes may be found in the distribution of the median nerve, most often impaired two-point discrimination, pinprick and light touch sensation, or occasionally hyperesthesia in the thumb, index, and middle fingers, with sparing of the skin over the thenar eminence. Thenar (abductor pollicis brevis muscle) weakness and atrophy may be present in advanced cases of CTS (Fig. 76.6). Flexing the patient’s hand at the wrist for 1 minute (Phalen maneuver) or hyperextension of the wrist (reversed Phalen maneuver) can reproduce the symptoms, is present in about 80% of patients, and is rarely false positive. A positive Tinel sign, in which percussion of the nerve at the carpal tunnel causes paresthesias in the distribution of the distal distribution of the median nerve, is present in approximately 60% of affected patients but is not specific for CTS and may be false positive.
Work-related wrist and hand symptoms (repetitive motion injury) from cumulative trauma in the workplace have received increasing attention by the general public in recent years (Thomsen et al., 2002). Although a proportion of these cases have bona fide CTS, longitudinal natural history data suggest that the majority of industrial workers do not develop symptoms of CTS (Nathan et al., 1998). Symptoms consistent with hand and wrist arthritis in a variety of occupational settings are now recognized as being much more common than CTS (Dillon et al., 2002). CTS appears to occur in work settings that include repetitive forceful grasping or pinching, awkward positions of the hand and wrist, direct pressure over the carpal tunnel, and the use of handheld vibrating tools. Increased risk for the syndrome has been found in meat packers, garment workers, butchers, grocery checkers, electronic assembly workers, musicians, dental hygienists, and housekeepers. The highest reported incidence of work-related CTS, based on the number of carpal tunnel surgeries performed, was 15% among a group of meat packers. Although computer keyboard use has long been thought to be related to developing carpal tunnel symptoms, recent data provide no convincing correlation between intensive keyboard use and the subsequent development of CTS (Papanicolaou et al., 2001; Stevens et al., 2001).
Diseases and conditions that have been found to predispose to the development of CTS include pregnancy, diabetes, obesity, age, rheumatoid arthritis, hypothyroidism, amyloidosis, gout, acromegaly, certain mucopolysaccharidoses, arteriovenous shunts for hemodialysis, old fractures at the wrist, and inflammatory diseases involving tendons or connective tissues at the wrist level (Becker et al., 2002). On rare occasions, CTS may be familial, and some patients with CTS have carpal canals that are significantly narrower than average.
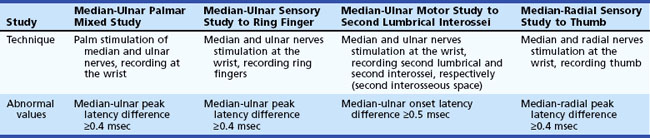
The most commonly performed EDX test for CTS are the median nerve sensory and motor conduction studies, which exhibit delayed sensory or motor latencies across the wrist in about 70% of patients. However, these studies are not sensitive enough in the diagnosis of CTS and fail to detect up to a third of patients with CTS, particularly those with mild and early symptoms. Recording the median latency at short distances over the course of the median nerve from palm to wrist and/or comparing this latency with the latency for the ulnar or radial nerve at the same distance (internal comparison nerve conduction studies) increase the sensitivity of these nerve conduction studies (Stevens, 1997) (Table 76.5).
In cases with only mild sensory symptoms, treatment with splints in neutral position, nonsteroidal antiinflammatory drugs (NSAIDs), and local corticosteroid injection often suffice. Withdrawal of provoking factors is also important. Although nonoperative treatments have been advocated (Osterman et al., 2002), a comparison of splinting versus surgery suggested that the latter may have a better long-term outcome than the former (Gerritsen et al., 2002). Use of a range of devices and appliances to protect the hand against CTS, including gel-padded gloves, has shown little if any improvement in objective measures of nerve function. Severe sensory loss and thenar atrophy suggest the need for surgical carpal tunnel release. Open surgical sectioning of the volar carpal ligament or fiberoptic techniques are often successful, with more than 90% of patients having prompt resolution of pain and paresthesias (Mirza and King, 1996). Improvement in distal latencies may lag behind the relief of symptoms. Comparing with preoperative values, nerve conduction studies demonstrate improvement in those with moderate abnormalities preoperatively, whereas patients with severe or no abnormalities on baseline nerve conduction studies had poorer results (Bland, 2001). A correlation between patients seeking workers’ compensation who hire attorneys and poorer operative outcomes has also been reported (Katz et al., 2001a). Older individuals may not improve as much as younger patients (Porter et al., 2002), and factors such as poor mental health, significant alcohol consumption, longer disease duration, and male gender also portend a poorer outcome. Rarely, symptoms persist after operation. Poor surgical results usually are associated with incomplete sectioning of the transverse ligament, surgical damage of the palmar cutaneous branch of the median nerve by an improperly placed skin incision, scarring within the carpal tunnel, or an incorrect preoperative diagnosis. Surgical reexploration may be required in diagnostically certain cases with poor response to the initial operation (Steyers, 2002).
Ulnar Nerve
Ulnar Nerve Entrapment at the Elbow
Ulnar mononeuropathy is the second most common entrapment or compression mononeuropathy, although it is considerably less common than CTS. Compression of the ulnar nerve by a thickened, fibrotic flexor carpi ulnaris aponeurosis (humeral-ulnar aponeurosis) at the entrance of the elbow’s cubital tunnel is a common cause of ulnar neuropathy (cubital tunnel syndrome). Patients with a subluxed ulnar nerve are at high risk for compression at the elbow. Also, prolonged and frequent resting of the flexed elbow on a hard surface such as a desk or armchair may result in external pressure to the nerve (ulnar groove syndrome). Occupations involving repeated flexion of the elbow may on occasion cause symptoms of ulnar neuropathy. A flexed elbow position increases both the intraneural and extraneural pressure on the nerve. The nerve at the site of repeated compression is associated with fibrous thickening, when a spindle-shaped swelling can often be felt. Other possible sources of injury of the ulnar nerve at the elbow include direct compression when the patient uses the arms to raise up in bed following surgical operations (Stewart and Shantz, 2003) or after periods of prolonged unconsciousness. The ulnar nerve at the elbow may be acutely injured as a result of fracture or dislocation involving the lower end of the humerus and the elbow joint. Occasionally, however, the nerve becomes chronically compressed years after such an injury, which often has led to cubitus valgus deformity (“tardy ulnar palsy”). The nerve may be damaged by osteophyte outgrowths resulting from arthritis of the elbow joint, by a ganglion or lipoma, by a Charcot elbow, and by the epitrochleoanconeus muscle and/or its dense fibrotendinous band. The ulnar nerve may also be involved in conditions that are known to increase the susceptibility of nerves to compression, such as diabetes mellitus or HNPP. Ulnar neuropathy at the elbow segment may also occur without any apparent cause.
Compared to evaluating entrapment neuropathies such as CTS, the EDX studies used to confirm and localize ulnar nerve entrapment at the elbow are more challenging. Localizing ulnar neuropathy at the elbow relies upon the demonstration of focal demyelination across the elbow, namely slowed motor conduction velocity (>10 meters per second) or conduction block (localized reduction in CMAP amplitude and area of >20%-30%) or both. Focal slowing or conduction block may be found in the elbow segment in more than 75% of cases (Azrieli et al., 2003). Performing additional ulnar motor conduction study, recording the first dorsal interosseous muscle in addition to recording the abductor digiti minimi muscle, increases the yield of finding focal slowing or conduction block. In the remaining patients, localization becomes less precise because of predominant axonal loss. To provide the extra nerve length needed during elbow flexion, the ulnar nerve is anatomically redundant in the ulnar groove when the elbow is extended, and this can cause measurement errors. A flexed position of the elbow (70 to 90 degrees) is preferred to the extended position when doing ulnar motor conduction studies to localize an ulnar lesion at the elbow. Short-segment incremental studies (“inching”) by stimulating the ulnar nerve in successive 1-cm increments across the elbow, looking for either an abrupt drop in amplitude or increase in latency, is a useful technique that helps to precisely localize the ulnar nerve lesion. Electrophysiological tests are helpful in differentiating between an ulnar neuropathy and a C8 nerve root or brachial plexus lesion. Ulnar sparing in ulnar sensory studies points to C8 radiculopathy, and needle EMG of C8 muscles innervated by the median nerve (e.g., abductor pollicis brevis, flexor pollicis longus) and radial nerve (e.g., extensor indicis proprius) helps exclude a C8 root lesion or a lower brachial plexopathy. MRI of the elbow may reveal a space-occupying lesion or anomalous structures impinging on the nerve or demonstrate nerve enlargement and increased signal intensity, even in the absence of localizing electrophysiological abnormalities (Vucic et al., 2006). High-resolution sonography at the elbow is also reported to accurately detect thickening of the ulnar nerve at the elbow (Beekman et al., 2004).
Conservative treatment should be attempted in patients with mild or intermittent sensory symptoms or in those with symptoms brought on by occupational causes. Avoidance of repetitive elbow flexion and extension or direct pressure on the elbow may alleviate the symptoms. Elbow protectors are helpful in patients with a history of excessive elbow leaning. Conservative treatment should be continued for at least 3 months before surgery is considered. Several surgical approaches to an ulnar nerve lesion at the elbow are possible, each with its proponents and critics. Techniques include simple release of the flexor carpi ulnaris aponeurosis, anterior transposition of the nerve trunk, and resection of the medial epicondyle. The choice of procedure should be tailored to the specific lesion found at surgery and may be assisted by short-segment incremental electrophysiological studies (“inching”). Transposition of the nerve trunk carries a higher rate of complications than ulnar neurolysis (Biggs and Curtis, 2006). Depending on the type of surgery and the severity and duration of neuropathy, response to these procedures will vary. Only about 60% of patients, especially those with symptoms of less than 1 year’s duration, benefit from surgery; some experience worsening of symptoms. It appears that those with more thickening of the nerve at the time of diagnosis (as determined by sonography) have a more unfavorable outcome, and those with electrophysiological signs of demyelination across the elbow have a more favorable course (Beekman et al., 2004).




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


