Chapter 79 Disorders of Skeletal Muscle
Striated muscle is the tissue that converts chemical energy into mechanical energy. The component processes include (1) excitation and contraction occurring in the muscle membranes, (2) the contractile mechanism itself, (3) various structural supporting elements that allow the muscle to withstand the mechanical stresses, and (4) the energy system that supports the activity and integrity of the other three systems. The logical categorization of myopathies is according to the part of the system involved. Until recently, this was impossible because the molecular basis of muscle activity was unknown. Scientific advances since the 1980s made this classification possible. Abnormalities in the membrane ion channels (channelopathies) involved in muscle excitation cause various forms of myotonia and periodic paralysis (see Chapter 64). The complex of proteins that include dystrophin, the sarcoglycans, and α-laminin constitute a vital structural mechanism linking the contractile proteins with the extracellular supporting structures. Defects in these proteins are the basis of many forms of muscular dystrophy. Although knowledge remains incomplete, it seems reasonable to modify the classic description of the myopathies to incorporate the new information. For this reason, in the sections that follow, disease descriptions are under the heading of their known molecular defect where possible; the classic appellation appears parenthetically. Before describing the illnesses themselves, we first review the techniques used in the clinical evaluation of patients.
Muscle Histology
The technique of muscle biopsy is not difficult. Under local anesthesia, a small incision made over the muscle allows, with careful dissection, removal of a small strip of muscle. Needle biopsies are useful in some situations. Histochemical studies of frozen sections are essential for proper interpretation. A transverse section of normal muscle shows fibers that are roughly of equal size and average approximately 60 mm in transverse diameter (Fig. 79.1). The muscle fibers of infants and young children are proportionately smaller. Each fiber consists of hundreds of myofibrils separated by an intermyofibrillar network containing aqueous sarcoplasm, mitochondria, and the sarcoplasmic reticulum with the associated transverse tubular system. Surrounding each muscle fiber is a thin layer of connective tissue (the endomysium). Strands of connective tissue group fibers into a fascicle, separated from each other by the perimysium. Groups of fascicles are collected into muscle bellies surrounded by epimysium.
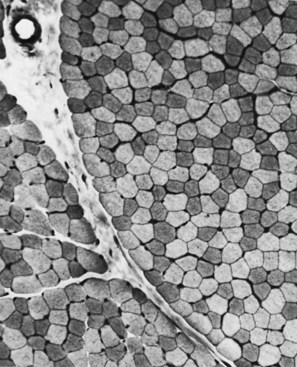
Situated at the periphery of the fibers are the sarcolemmal nuclei. The fibers are of different types. The simplest division is into type 1 and type 2 fibers, best demonstrated with the histochemical reaction for myosin adenosine triphosphatase (ATPase) (Fig. 79.2). The type 1 and type 2 fibers are roughly equivalent to slow and fast fibers or to oxidative and glycolytic fibers in human muscle. The best demonstration of the intermyofibrillar network pattern is with the histochemical reactions for oxidative enzymes, such as reduced nicotinamide adenine dinucleotide dehydrogenase. A regular network extends across the whole fiber. In addition to the routine stains with hematoxylin and eosin, modified Gomori-trichrome, myosin ATPase, and nicotinamide adenine dinucleotide dehydrogenase, the use of other special stains demonstrates fat (Sudan black or oil red 0), complex carbohydrates (periodic acid–Schiff), amyloid (Congo red), or specific enzymes (e.g., phosphorylase, succinic dehydrogenase, cytochrome oxidase). Immunocytochemical techniques demonstrate the location and integrity of structural proteins such as dystrophin. They also characterize cell types in biopsy samples with inflammatory changes.
Changes of Denervation
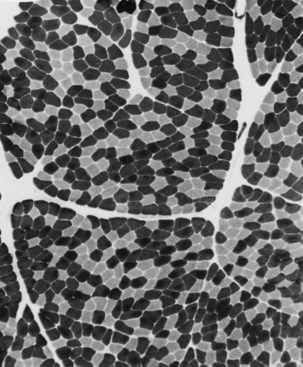
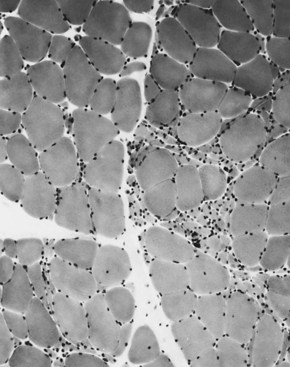
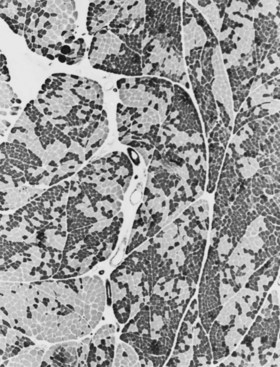
When muscle loses its nerve supply, muscle fibers atrophy, often resulting in fiber squeezing into the spaces between normal fibers and assuming an angulated appearance (Fig. 79.3). Scattered angulated fibers appear early in denervation. Sometimes, picturesque changes in the intermyofibrillar network occur, as in the target fiber, which characterizes denervation and reinnervation. This is a three-zone fiber on which the intermediate zone stains more darkly, and the central “bull’s eye” stains much lighter than normal tissue (Fig. 79.4). Often a neighboring nerve twig reinnervates a denervated fiber. This results in the same anterior horn cell supplying two or more contiguous fibers. If that nerve twig then undergoes degeneration, instead of only one small angulated fiber being produced, a small group of atrophic fibers develops. Group atrophy suggests denervation (Fig. 79.5). As the process continues, large groups of geographical atrophy occur in which entire fascicles are atrophic. In addition to the change in size, a redistribution of the fiber types occurs as well. Normally a random distribution of type 1 and 2 muscle fiber types exists, sometimes incorrectly called a checkerboard or mosaic pattern. The same process of denervation and reinnervation results in larger and larger groups of contiguous fibers supplied by the same nerve. Because all fibers supplied by the same nerve are of the same fiber type, groups of type 1 fibers next to groups of type 2 fibers replace the normal random pattern. This fiber type grouping is pathognomonic of reinnervation (Fig. 79.6). When long-standing denervation is present, the atrophic muscle fibers almost disappear, leaving small clumps of pyknotic nuclei in their place.
Myopathic Changes
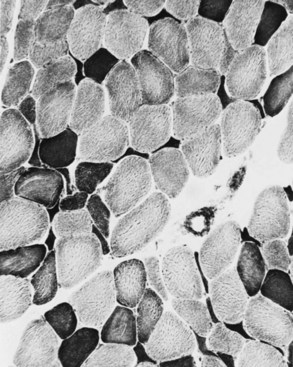
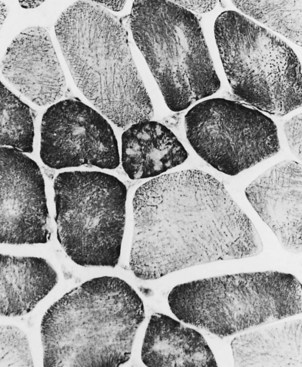
Changes in the intermyofibrillar network pattern are common in myopathic disorders. There is often a moth-eaten, whorled change to the intermyofibrillar network in LGMD and facioscapulohumeral dystrophy (FSHD) (Fig. 79.7); the intermyofibrillar network loses its orderly arrangement and swirls, resembling the current in an eddying stream. These changes may be seen in several diseases but tend to be much more common in the myopathies.
Other Changes
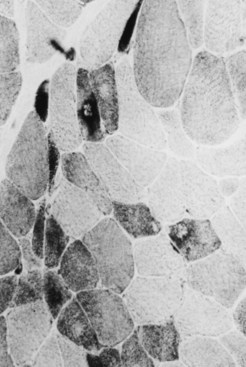
Selective changes in fiber types occur. Type 2 fiber atrophy is one of the most common abnormalities seen in muscle (Fig. 79.8). Type 2 atrophy, particularly if limited to type 2B fibers, is nonspecific and indicates muscle disuse. If a limb is casted and the muscle examined some weeks later, selective atrophy of type 2 fibers is noted. Any chronic systemic illness tends to produce type 2 atrophy. It occurs in rheumatoid arthritis, nonspecific collagen vascular diseases, cancer (hence the name cachectic atrophy), mental retardation in children, and pyramidal tract disease. Type 2B fiber atrophy can also result from chronic corticosteroid administration. Therefore, type 2 fiber atrophy should probably be regarded as a nonspecific result of anything less than robust good health.

Specific Disorders
Muscular Dystrophies
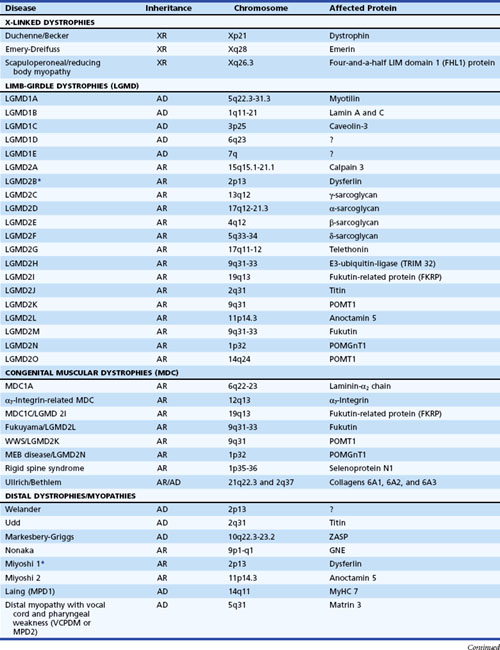
The muscular dystrophies are a group of hereditary muscle disorders that occur at all ages and with varying degrees of severity. The traditional classification is on clinical grounds. Increasing information about the molecular basis of these disorders provides both reassurance and puzzlement to clinicians (Table 79.1). Different dystrophies are due to distinct molecular abnormalities; however, patients with similar molecular defects may show a wide variability in phenotype not always easily explained.
Dystrophin Deficiency (Duchenne Muscular Dystrophy, Becker Muscular Dystrophy, and Atypical Forms)
An absence or deficiency of dystrophin is responsible for two disorders that cause progressive destruction of muscle. The responsible gene is located on the short arm of the X chromosome at locus Xp21. The gene is extremely large, comprising more than 2.5 million base pairs and 79 exons or coding regions. Approximately two-thirds of cases are associated with a detectable deletion or duplication of segments within the gene. The others are presumably due to point mutations too small to be detected using standard techniques. “Hot spots” for these gene deletions exist, notably between exons 43 and 52 and particularly 44 and 49 (Nobile et al., 1997). Whether the deletion is in frame or out of frame (see Chapter 40) determines whether dystrophin is absent from the muscle or present in a reduced altered form. This has clinical significance because the former is usually associated with the severe Duchenne variety of the disease (DMD), whereas the latter may cause the milder Becker variant (BMD). In BMD, the abnormal dystrophin preserves enough function to slow down the progress of the illness. Reading of the DNA code is triplet by triplet. Maintenance of this reading frame throughout the length of the gene is required for dystrophin production. If a deletion removes a multiple of three base pairs, the reading frame may be intact upstream and downstream and may make limited sense, as if the sentence “You cannot eat the cat” were changed to “You not eat the cat,” and some modified dystrophin may be formed. This is often the situation in the mild form of dystrophin deficiency. In the severe form, the reading frame is destroyed, as if a deletion resulted in the sentence “Yoc ann ote att hec at.” Exceptions to this rule exist, and frameshift deletions have been associated with the milder form of the disease, particularly at the 5′ end of the gene in exons 3 to 7.
Duchenne Muscular Dystrophy
Even in the severe variety of DMD, affected children are normal at birth. During the second year when the boys begin walking, the clumsiness seen in all toddlers persists. Soon the children have to place one hand on the knee to assume an upright position when rising from the floor (Gower maneuver). Often at this stage, the calf muscles are rather firm and rubbery (pseudohypertrophy) (Fig. 79.9). Within 2 to 3 years, parents notice that the child runs properly and is never able to jump clear of the floor with both feet. In the absence of therapy, tightness across several joints in the legs is noted. The iliotibial bands and the heel cords are usually the first to become tight. This is particularly noticeable in boys who habitually walk on their toes.

Fig. 79.9 Duchenne muscular dystrophy. Calf and thigh hypertrophy in an ambulatory 8-year-old patient.
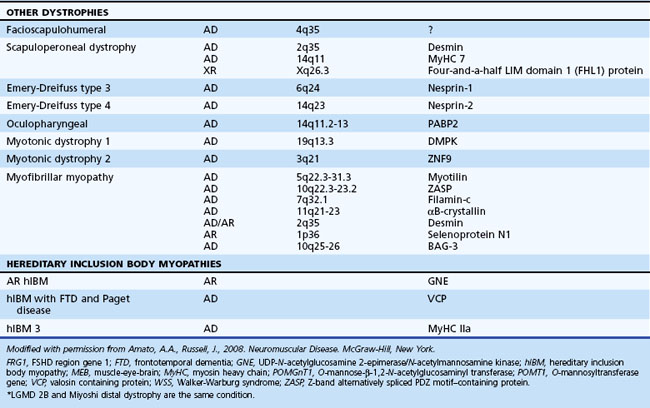
The serum concentration of creatine kinase (CK) is markedly elevated in this illness; levels greater than 10,000 mU/mL are common. Electromyography (EMG) shows myopathic changes (see Chapter 32B), and muscle biopsy demonstrates variation in the size of fibers, fibrosis, groups of basophilic fibers, and opaque or hypercontracted fibers (hyaline fibers) (Figs. 79.10 to 79.12).
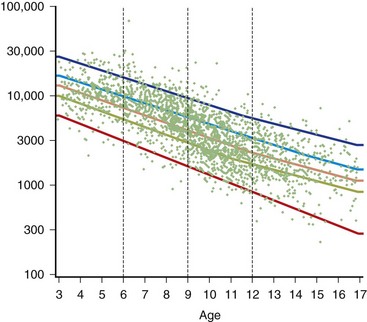
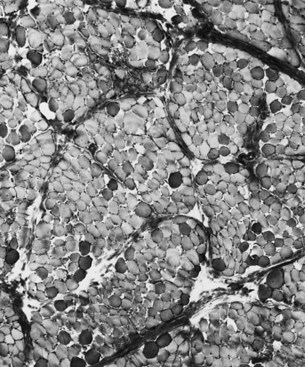
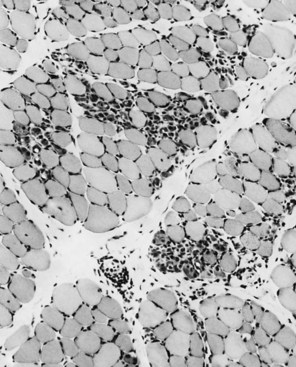
Genetic Counseling
Genetic analysis of all potential carriers is advisable. In a family in which the disease is associated with a deletion, there is little problem in determining whether the woman is carrying the affected X chromosome, using techniques that are presently available. Genetics laboratories now have the ability to identify the presence of a mutant gene over the background contributed by the normal allele. This involves an analysis of the gene “dosage,” comparing two normal alleles that have a double dose against a deleted allele and a normal allele that have a single dose (Voskova-Goldman et al., 1997). The basis for current diagnosis of carrier status after identification of a deletion in a proband is by analysis of a gene dosage. Our diagnostic strategy uses fluorescence in situ hybridization (FISH) to detect female carriers with major deletions in the dystrophin gene. Unfortunately, situations exist in which a mutation is identified in a boy with “sporadic” DMD but not in the mother, and yet the mother is still a carrier. This occurrence is secondary to germline mosaicism in which the mutation in the mother lies only in a percentage of her oocytes. The estimated recurrence rate of DMD is as high as 14% even in such cases. Prenatal diagnosis using amniotic cells or chorionic villus biopsies can identify the affected fetus and, more importantly, those who are unaffected.
Other Limb-Girdle Dystrophies
The most important recent development in muscle disease relates to a group of disorders that were clearly dystrophic but defied proper classification. The traditional diagnosis of LGMDs cloaked the clinician’s uncertainty. In some patients, weakness was generally proximal, but other characteristics were disparate. Some cases were dominantly inherited, others recessively. Some had more hip than shoulder weakness, others the reverse. The illness could be mild in late life, others severe and early. There was general recognition that the rubric LGMD encompassed a group of different illnesses. Beginning with the discovery that a defect in one of the sarcoglycans caused a severe form of dystrophy occurring in North Africa, the delineation of several other entities characterized by defects in structural proteins or enzymes followed. These include the sarcoglycans, the α2 chain of laminin (merosin), calcium-activated protease, calpain-3, and others. The designation of autosomal dominant LGMD is type 1, and the designation of autosomal recessive LGMD is type 2. Subclassification with an alphabetical letter characterizes distinct genetic forms of LGMD1 and LGMD2 (see Table 79.1). The following sections outline the known protein abnormalities and then comment on the more amorphous forms of LGMD. The prevalence of these diseases as a group probably approaches 1 per 100,000 (van der Kooi et al., 1996). The autosomal recessive LGMDs are more common than the autosomal dominant LGMDs.
Autosomal Dominant Limb-Girdle Muscular Dystrophies
LGMD1A (Myotilin Deficiency)
LGMD1A is allelic to one subtype of myofibrillary myopathy and is caused by mutations in the myotilin gene located on chromosome 5q22.3-31.3 (Selcen and Engel, 2004). Myotilin is a sarcomeric protein that is present at the Z-disc. The protein is likely important in myofibrillogenesis and stabilization of the Z-disc and sarcomere.
LGMD1B (Lamin A/C Deficiency)
This myopathy is allelic with the disorder previously reported as autosomal dominant Emery-Dreifuss muscular dystrophy (EDMD) (Bonne et al., 2000; van der Kooi et al., 1996). Some patients manifest with a limb-girdle pattern of weakness, while others present with a humeral-peroneal weakness and early contractions. Cardiomyopathy with severe conduction defects and arrhythmias may also occur, with or without skeletal muscle involvement. Sudden death secondary to fatal arrhythmias is common, and pacemaker insertion is often necessary. Serum CK levels may be normal or elevated up to 25-fold. Muscle biopsies demonstrate dystrophic features with the rare occurrence of rimmed vacuoles.
LGMD1B is localized to mutations in the lamin A/C gene located on chromosome 1q11-21 (Bonne et al., 1999; van der Kooi et al., 1997). Alternative splicing of the lamin A/C messenger (m)RNA transcript produces lamins A and C. Lamin A/C is an intermediate-size filament located on the nucleoplasmic surface of the inner nuclear membrane, where it interacts with various lamin-associated proteins including emerin, the abnormal protein associated with X-linked EDMD. Lamin A/C may also bind to heterochromatin. Immunostaining of the nuclear membrane with anti-emerin antibodies is normal, helping distinguish this myopathy from X-linked EDMD. Electron microscopy reveals alterations in myonuclei, including the loss of peripheral heterochromatin, altered interchromatin texture, and fewer than normal nuclear pores (Sabatelli et al., 2001).




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


