Video 53.1). In contrast, patients with MG tend not to complain of “generalized weakness,” “generalized fatigue,” “sleepiness,” or muscle pain. In the classic case, fluctuating weakness is worse with exercise and improved with rest. Symptoms tend to progress later in the day. Many different factors can precipitate or aggravate weakness, such as physical stress, emotional stress, infection, or exposure to medications that impair neuromuscular transmission (perioperative succinylcholine, aminoglycoside antibiotics, quinine, quinidine, and botulinum toxin).
B. Diagnosis. The diagnosis is based on a history of fluctuating weakness with corroborating findings on examination (Video 53.2![]() ). There are several different ways to validate or confirm the clinical diagnosis.
). There are several different ways to validate or confirm the clinical diagnosis.
1. Edrophonium test. The most immediate and readily accessible confirmatory study is the edrophonium test. To perform the test, choose one or two weak muscles to judge. Eyelid ptosis, dysconjugate gaze, and other cranial deficits provide the most reliable endpoints. Use a setting where hypotension, syncope, or respiratory failure can be managed as patients occasionally decompensate during the test. If the patient has severe dyspnea, defer the test until the airway is secure. Start an intravenous (IV). Have IV atropine 0.4 mg readily available in the event of bradycardia or extreme gastrointestinal (GI) side effects. Edrophonium 10 mg (1 mL) is drawn up in a syringe, and 1 mg (0.1 mL) should be given as a test dose while checking the patient’s heart rate (to assure the patient is not supersensitive to the drug). If no untoward side effects occur after 1 minute, another 3 mg is given. Many MG patients will show improved power within 30 to 60 seconds of giving the initial 4 mg at which point the test can be stopped. If after 1 minute there is no improvement, give additional 3 mg, and if there is still no response, 1 minute later give the final 3 mg. If the patient develops muscarinic symptoms or signs at any time during the test (sweating, salivation, and GI symptoms), or should fasciculations be detected then one can assume that enough edrophonium has been given to see improvement in strength and the test can be stopped. When a placebo effect or examiner bias is of concern, the test is performed in a double-blind placebo control fashion. The 1 mL control syringe contains either saline, 0.4 mg atropine, or nicotinic acid 10 mg. Improved strength from edrophonium lasts for just a few minutes. When improvement is clear-cut, the test is positive. If the improvement is borderline, it is best to consider the test negative. The test can be repeated several times. Sensitivity of the edrophonium test is about 90%. The specificity is difficult to determine because improvement following IV edrophonium has been reported in other neuromuscular diseases including Lambert–Eaton myasthenic syndrome (LEMS), botulism, Guillain–Barré syndrome (GBS), motor neuron disease, and with lesions of the brainstem, pituitary, and cavernous sinus. Neostigmine has a longer duration of effect and in selected patients may be an alternative cholinesterase inhibitor (CEI) for diagnostic testing, especially in children. For performance of a “neostigmine test,” 0.04 mg/kg is given intramuscularly or 0.02 mg/kg intravenously (one time only).
2. ACh receptor antibodies. The primary serologic test is the immunoprecipitation assay for ACh receptor binding antibodies. In addition, assays for receptor modulating and blocking antibodies are available. Binding antibodies are present in about 80% of all MG patients (40% to 50% of patients with pure ocular MG, 80% of those with mild generalized MG, and 90% of patients with moderate to severe generalized MG, and 70% of those in clinical remission). By also testing for modulating and blocking antibodies, the sensitivity improves to 90% overall. Specificity is superb with false positives exceedingly rare in reliable labs.
3. MuSK antibodies. Approximately 25% of patients seronegative for ACh receptor antibodies have antibodies to muscle-specific kinase (MuSK). The clinical features of MuSK positive patients may differ from non-MuSK MG patients. Such patients tend to be younger women (under age 40) with disproportionate bulbar, neck extensor, shoulder, and respiratory symptoms with increased likelihood of “fixed weakness” and have a lower likelihood of abnormal repetitive stimulation and edrophonium test results. MuSK patients have no associated thymus abnormalities and are more likely to be refractory to conventional medical management.
4. LRP4 (low-density lipoprotein receptor-related protein 4) and agrin antibodies present in a small number of patients.
5. EMG (electrophysiological testing). Repetitive stimulation testing is widely available and has variable sensitivity depending on the number and selection of muscles studied and various provocative maneuvers. However, in most laboratories, this technique has a sensitivity of about 50% in all patients with MG (lower in patients with mild or pure ocular disease). In general, the yield from repetitive stimulation is higher when testing muscle groups having clinically significant weakness. Single-fiber EMG (SFEMG) is a highly specialized technique, usually available in major academic centers, with a sensitivity of about 90%. Abnormal single-fiber results are common in other neuromuscular diseases; therefore, the test must be used in the correct clinical context. The specificity of SFEMG is an important issue in that mild abnormalities can clearly be present with a variety of other diseases of the motor unit including motor neuron disease, peripheral neuropathy, and myopathy. Disorders of neuromuscular transmission other than MG can have abnormalities on SFEMG. In contrast, ACh receptor antibodies (and MuSK antibodies) are not found in non-MG patients. In summary, the two highly sensitive laboratory studies are SFEMG and ACh receptor antibodies; nonetheless, neither test is 100% sensitive.
C. Prognosis. Natural course: Appropriate management of the patient with autoimmune MG requires understanding of the natural course of the disease. The long-term natural course of MG is highly variable but generalizations are as follows. About half of MG patients present with ocular symptoms (ptosis or diplopia) and by 1 month 80% have eye findings. The presenting weakness is bulbar in 10%, limb in 10%, generalized in 10%, and respiratory in 1%. By 1 month, symptoms remain purely ocular in 40%, generalized in 40%, limited to the limbs in 10%, and limited to bulbar muscles in 10%. Weakness remains restricted to the ocular muscles on a long-term basis in about 15% to 20% (pure ocular MG). Most patients with initial ocular involvement tend to develop generalized weakness within the first year of the disease (90% of those who generalize do so within the initial 12 months). Maximal weakness occurs within the initial 3 years in 70% of patients. In the modem era, death from MG is rare. Spontaneous long-lasting remission occurs in about 10% to 15%, usually in the first or second year of the disease. Most MG patients develop progression of clinical symptoms during the initial 2 to 3 years. However, progression is not uniform, as illustrated by 15% to 20% of patients whose symptoms remain purely ocular and those who have spontaneous remission.
D. Treatment.
1. First-line therapy. CEIs are safe, effective, and first-line therapy in all patients. Inhibition of acetylcholinesterase (AChE) reduces the hydrolysis of ACh, increasing the accumulation of ACh at the nicotinic postsynaptic membrane. The CEIs used in MG bind reversibly (as opposed to organophosphate CEIs, which bind irreversibly) to AChE. These drugs cross the blood–brain barrier poorly and tend not to cause central nervous system (CNS) side effects. Absorption from the GI tract tends to be inefficient and variable, with oral bioavailability of about 10%. Muscarinic autonomic side effects of GI cramping, diarrhea, salivation, lacrimation, and diaphoresis are common and dose-related, and occasional patients may have bradycardia. A feared potential complication of excessive CEI use is skeletal muscle weakness (cholinergic weakness). Patients receiving parenteral CEI are at the greatest risk to have cholinergic weakness. It is uncommon for patients receiving oral CEI to develop significant cholinergic weakness even while experiencing muscarinic cholinergic side effects. Generally available CEIs are summarized in Table 53.1.

a. Pyridostigmine (Mestinon) is the most widely used CEI for long-term oral therapy. Onset of effect is within 30 minutes of an oral dose, with peak effect within 1 to 2 hours, and wearing off gradually at 3 to 4 hours postdose. The starting dose is 30 to 60 mg three to four times per day depending on symptoms. Optimal benefit usually occurs with a dose of 60 mg every 4 hours. Muscarinic cholinergic side effects are common with larger doses. Patients with significant bulbar weakness will often time their dose about 1 hour before meals in order to maximize chewing and swallowing. Of all the CEI preparations, pyridostigmine has the least muscarinic side effects. Pyridostigmine may be used in a number of alternative forms to the 60 mg tablet. The syrup may be necessary for children or for patients with difficulty swallowing pills. Sustained-release pyridostigmine 180 mg (Mestinon Timespan) is sometimes preferred for night time use. Unpredictable release and absorption limit its use. Patients with severe dysphagia or those undergoing surgical procedures may need parenteral CEI. IV pyridostigmine should be given at about 1/30 of the oral dose. Neostigmine (prostigmine) has a slightly shorter duration of action and somewhat greater muscarinic side effects.
b. For patients with intolerable muscarinic side effects at CEI doses required for optimal power, a concomitant anticholinergic drug such as atropine sulfate (0.4 to 0.5 mg orally) or glycopyrrolate (Robinul) (1 to 2 mg orally) on a p.r.n. basis or with each dose of CEI may prevent the side effects. Patients with mild disease can often be managed adequately with CEIs. However, patients with moderate, severe, or progressive disease will usually require more effective therapy.
2. Thymectomy. Association of the thymus gland with MG was first noted around 1900 and thymectomy has become standard therapy for over 50 years. A large randomized international multicenter controlled trial demonstrated that thymectomy improved clinical outcomes over a 3 year period in patients with nonthymomatous MG. The general consensus is that thymectomy should be considered for patients with moderate to severe MG, especially those inadequately controlled on CEI, and those under age 55 years. All patients with suspected thymoma undergo surgery. About 75% of MG patients appear to benefit from thymectomy. Patients may improve or simply stabilize. For unclear reasons, the onset of improvement tends to be delayed by a year or two in most patients (some patients may improve 5 to 10 years after surgery). The majority of centers use the transsternal approach for thymectomy with the goal of complete removal of the gland. The limited transcervical approach has been largely abandoned due to the likelihood of incomplete gland removal. Many centers perform a “maximal thymectomy” in order to ensure complete removal. The procedure involves a combined transternal–transcervical exposure with en bloc removal of the thymus. Thorascopic and video-assisted thymectomy offer less invasive options. If thymectomy is to be performed, choose an experienced surgeon, anesthesiologist, and a medical center with a good track record and insist that the entire gland is removed.
a. Which patients do not undergo thymectomy? Patients with very mild or trivial symptoms do not have surgery. Most patients with pure ocular MG do not undergo thymectomy even though there has been some reported benefit in selected patients. Thymectomy is often avoided in children due to the theoretical possibility of impairing the developing immune system. However, reports of thymectomy in children as young as 2 to 3 years of age have shown favorable results without adverse effects on the immune system. Thymectomy has been largely discouraged in patients over age 55 because of expected increased morbidity, latency of clinical benefit, and frequent observation of an atrophic, involuted gland. Major complications from thymectomy are uncommon so long as the surgery is performed at an experienced center with anesthesiologists and neurologists familiar with the disease and perioperative management of MG patients. Common, though less serious, aspects of thymectomy include postoperative chest pain (which may last several weeks), a 4- to 6-week convalescence period, and cosmetically displeasing incisional scar.
3. Corticosteroids. There are no controlled trials documenting the benefit of corticosteroids in MG. However, nearly all authorities have personal experience attesting to the virtues (and complications) of corticosteroid use in MG patients. In general, corticosteroids are used in patients with moderate to severe disabling symptoms that are refractory to CEI. Patients are commonly hospitalized to initiate therapy due to the risk of early exacerbation. Opinions differ regarding the best method of administration. For patients with severe MG it is best to begin with high-dose daily therapy of 60 to 80 mg/day orally. Early exacerbation occurs in about half of patients, usually within the first few days of therapy and typically lasting 3 or 4 days. In 10% of cases, the exacerbation is severe, requiring mechanical ventilation or a feeding tube (thus, the need to initiate therapy in the hospital). Overall, about 80% of patients show a favorable response to steroids (with 30% attaining remission and 50% having marked improvement). Mild to moderate improvement occurs in 15%, and 5% have no response. Improvement begins as early as 12 hours and as late as 60 days after beginning prednisone, but usually the patient begins to improve within the first or second week. Improvement is gradual, with marked improvement occurring at a mean of 3 months, and maximal improvement at a mean of 9 months. Of those patients having a favorable response, most maintain their improvement with gradual dosage reduction at a rate of 10 mg every 1 to 2 months. More rapid reduction is usually associated with a flare-up of the disease. Although many patients can eventually be weaned off steroids and maintain their response, the majority cannot. They require a minimum dose (5 to 30 mg alternate day [AD]) in order to maintain their improvement. Complications of long-term high-dose prednisone therapy are substantial, including cushingoid appearance, hypertension, osteoporosis, diabetes, cataracts, aseptic necrosis, and the other well-known complications of chronic steroid therapy. Older patients tend to respond more favorably to prednisone. An alternative prednisone regimen involves low-dose AD gradually increasing schedule in an attempt to avoid the early exacerbation. Patients receive prednisone 25 mg AD with increase by 12.5 mg every third dose (about every fifth day) to a maximum dose of 100 mg AD or until sufficient improvement occurs. Clinical improvement usually begins within 1 month of treatment. The frequency and severity of early exacerbation is less than that associated with high-dose daily regimens. High-dose IV methylprednisolone (1,000 mg IV daily for 3 to 5 days) can provide improvement within 1 to 2 weeks but the clinical improvement is temporary.
4. Alternative immunosuppressive drug therapy.
a. Mycophenolate mofetil (CellCept) is a purine inhibitor widely used in recent years for the treatment of MG. Anecdotal uncontrolled experience would suggest that about 75% of MG patients benefit from the drug with the typical onset of improvement within 2 to 3 months. The drug is generally well tolerated. Typically begin with 250 to 500 mg orally twice a day, and over 2 to 4 weeks increase the dose to 1,000 mg orally twice a day. Two recently completed prospective controlled double-blind trials failed to demonstrate a benefit from mycophenolate in selected MG patients leading to a reevaluation of its role and suggesting that the anecdotal reports of benefit may be incorrect. Complications are uncommon and include GI intolerance and occasional patients with hepatic or hematologic abnormalities.
b. Azathioprine (Imuran) is a cytotoxic purine analog with extensive use in MG (but largely uncontrolled and retrospective). The starting dose is 50 mg by mouth daily, with complete blood count (CBC) and liver function tests weekly in the beginning. If the drug is tolerated and if the blood work is stable, the dose is increased by 50 mg every 1 to 2 weeks aiming for a total daily dose or about 2 to 3 mg/kg/day (about 150 mg/day in the average-sized adult). When azathioprine is first started, about 15% of patients will have intolerable GI side effects (nausea, anorexia, and abdominal discomfort) and sometimes associated with fever, leading to discontinuation. Bone marrow suppression with relative leukopenia (white blood cells (WBCs) 2,500 to 4,000) occurs in 25% of patients but is usually not significant. If the WBC count drops below 2,500 or the absolute granulocyte count goes below 1,000, the drug is stopped (and the abnormalities usually resolve). Macrocytosis is common and of unclear clinical significance. Liver enzymes elevate in 5% to 10% but are usually reversible, and severe hepatic toxicity occurs in only about 1%. Infection occurs in about 5%. There is a theoretical risk of malignancy (based on observations in organ transplant patients), but this increased risk has not been clearly established in the MG patient population. About half of MG patients improve on azathioprine with onset about 4 to 8 months into treatment. Maximal improvement takes about 12 months. Relapse after discontinuation of azathioprine occurs in over half of patients, usually within 1 year.
c. Cyclosporine is used in patients with severe MG who cannot be adequately managed with corticosteroids or azathioprine. The starting dose is 3 to 5 mg/kg/day given in two divided doses. Cyclosporine blood levels should be measured monthly (aiming for a level of 200 to 300) along with electrolytes, magnesium, and renal function (in general, serum creatinine should not exceed one and one-half times the pretreatment level). Blood should be sampled before the morning dose is taken. Over half of patients improve on cyclosporine. The onset of clinical improvement occurs about 1 to 2 months after beginning therapy, and maximal improvement occurs at about 3 to 4 months. Side effects include renal toxicity and hypertension. Nonsteroidal anti-inflammatory drugs (NSAIDs) and potassium-sparing diuretics are among the list of drugs that should be avoided while on cyclosporine. In patients on corticosteroids, the addition of cyclosporine can lead to a reduction in steroid dosage (although it is usually not possible to discontinue prednisone).
d. Methotrexate has been used in selected patients for decades with clinical response the subject of sporadic anecdotal reports.
e. Tacrolimus has been reported to be beneficial in several series and in some parts of the world is among the more commonly prescribed immunosuppressive options.
f. Rituximab has been reported to be effective in treating MG in selected patients (the anecdotal reports tend to involve relatively refractory patients who have done poorly with alternative treatment options). The anecdotal reports of rituximab benefits in MuSK patients are particularly notable given the disproportionate tendency for such patients to be refractory to many other immunosuppressive options.
g. Eculizumab has been reported to be effective in treating MG and is completing a large randomized trial. This monoclonal antibody blocks C5 and ostensibly reduces compliment mediated lysis of the postsynaptic membrane.
5. Plasma exchange (PLEX or plasmapheresis) removes ACh receptor antibodies and results in rapid clinical improvement. The standard course involves removal of 2 to 3 L of plasma every other day or 3 times per week until the patient improves (usually a total of three to five exchanges). Improvement begins after the first few exchanges and reaches the maximum within 2 to 3 weeks. The improvement is moderate to marked in nearly all patients but usually wears off after 4 to 8 weeks due to the reaccumulation of pathogenic antibodies. Vascular access may require placement of a central line. Complications include hypotension, bradycardia, electrolyte imbalance, hemolysis, infection, and access problems (such as pneumothorax from placement of a central line). Indications for PLEX include any patient in whom a rapid temporary clinical improvement is needed. Patients with MuSK antibodies who are refractory to other modalities may respond favorably to PLEX.
6. High-dose IV and subcutaneous immunoglobulin (IVIG) administration is associated with rapid improvement in MG symptoms in a time frame similar to plasma exchange. The mechanism is unclear but may relate to down-regulation of ACh receptor antibody production or to the effect of anti-idiotype antibodies. The usual protocol is 2 g/kg spread out over 5 consecutive days (0.4 g/kg/day). Different IVIG preparations are administered IV at different rates (contact the pharmacy for guidelines). The majority of MG patients improve, usually within 1 week of starting IVIG. The degree of response is variable and the duration of response is limited, as with plasma exchange, to about 4 to 8 weeks. Complications include fever, chills, and headache, which respond to slowing down the rate of the infusion and giving diphenhydramine. Occasional cases of aseptic meningitis, renal failure, nephrotic syndrome, and stroke have been reported. Also, patients with selective IgA deficiency can have anaphylaxis, best avoided by screening for IgA deficiency ahead of time. The treatment is relatively expensive, comparable to PLEX. In patients with problematic IV access or those using IG for long-term maintenance therapy a subcutaneous IG preparation is an alternative.
E. General guidelines for management.
1. Be certain of the diagnosis.
2. Patient education. Provide the patient with information about the natural course of the disease (including the variable and somewhat unpredictable course). Briefly review the treatment options outlined above pointing out effectiveness, time course of improvement, duration of response, and complications. Provide the patient with educational pamphlets prepared by the Myasthenia Gravis Foundation or the Muscular Dystrophy Association.
3. When to hospitalize the patient. Patients with severe MG can deteriorate rapidly over a period of hours. Therefore, those having dyspnea should be hospitalized immediately in a constant observation or intensive care setting. Patients with moderate or severe dysphagia, weight loss, as well as those with rapidly progressive or severe weakness should be admitted urgently. This will allow close monitoring and early intervention in the case of respiratory failure and will also expedite the diagnostic workup and initiation of therapy.
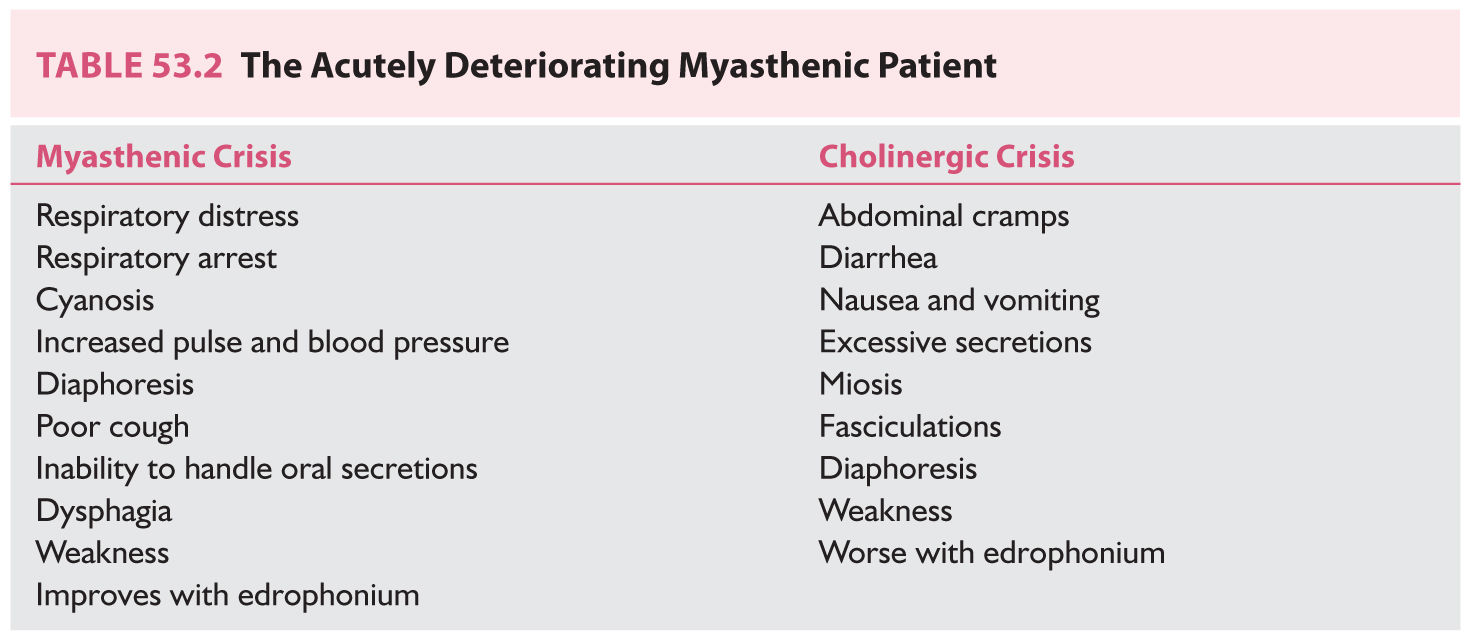
4. Myasthenic crisis (Table 53.2) is a medical emergency characterized by respiratory failure from diaphragm weakness or severe oropharyngeal weakness leading to aspiration. Crisis can occur in the setting of surgery (post-op), acute infection, or following rapid withdrawal of corticosteroids (though some patients have no precipitating factors). Patients should be placed in an intensive care unit (ICU) setting and have forced vital capacity (FVC) checked every 2 hours. Changes in arterial blood gases occur relatively late in neuromuscular respiratory failure. There should be a low threshold for intubation and mechanical ventilation. Criteria for intubation include a drop in the FVC below 15 mL/kg (or below 1 L in an average-sized adult), severe aspiration from oropharyngeal weakness, or labored breathing regardless of the measurements. If the diagnosis is not clear-cut, it is advisable to secure the airway with intubation, stabilize ventilation, and only then address the question of the underlying diagnosis. If the patient has been taking CEI, the drug should be temporarily discontinued in order to rule out the possibility of “cholinergic crisis.”
5. Screen for and correct any underlying medical problems such as systemic infection, metabolic problems (like diabetes), and thyroid disease (hypo or hyperthyroidism can exacerbate MG).
6. Drugs to avoid in MG. Avoid using d-penicillamine, α-interferon, chloroquine, quinine, quinidine, procainamide, and botulinum toxin. Aminoglycoside antibiotics should be avoided unless needed for a life-threatening infection. Fluoroquinolones (ciprofloxacin) and erythromycin have significant neuromuscular blocking effects. Telithromycin (Ketek) has been reported to cause life-threatening weakness in patients with MG and should not be used. Neuromuscular blocking drugs such as pancuronium and D-tubocurarine can produce marked and prolonged paralysis in MG patients. Depolarizing drugs such as succinylcholine can also have a prolonged effect and should be used by a skilled anesthesiologist who is well aware of the patient’s MG.
F. Guidelines for specific therapies. Treatment must be individualized. Mild diplopia and eyelid ptosis may not be disabling for some patients, but for a pilot or neurosurgeon, mild intermittent diplopia may be critical. In a similar fashion, some patients may tolerate side effects better than others.
1. Mild or trivial weakness, either localized or generalized, should be managed with a CEI such as pyridostigmine.
2. Moderate to marked weakness, localized or generalized, should initially be managed with a CEI. Even if symptoms are adequately controlled, patients under age 55 should be considered for thymectomy early in the course of the disease (within the first year). In older patients, thymectomy is usually not performed unless the patient is thought to have a thymoma. Thymectomy is performed at an experienced medical center with the clear intent of complete removal of the gland. All patients with suspected thymoma (by chest scan) should have thymectomy, even if their myasthenic symptoms are mild. Unless a thymoma is suspected, patients with pure ocular disease are usually not treated with thymectomy.
3. If symptoms are inadequately controlled on CEI, immunosuppression is used. High-dose corticosteroid therapy is the most predictable and effective long-term option. If patients have severe, rapidly progressive, or life-threatening symptoms, the decision to start corticosteroids is clear-cut. Patients with disabling but stable symptoms may instead receive a nonsteroidal immunosuppressive drug such as azathioprine.
4. PLEX or IVIG are indicated in:
a. Rapidly progressive, life-threatening, impending myasthenic crisis or actual crisis, particularly if prolonged intubation with mechanical ventilation is judged hazardous.
b. Preoperative stabilization of MG (such as prior to thymectomy or other elective surgery) in poorly controlled patients.
c. Disabling MG refractory to other therapies (maintenance therapy).
5. If these options fail, then use mycophenolate, cyclosporine, tacrolimus, or rituximab.
6. If the patient remains poorly controlled despite appropriate treatment, then perform a repeat chest computed tomography (CT) scan looking for residual thymus. Some patients improve after “repeat thymectomy.” Check for other medical problems (diabetes, thyroid disease, infection, and coexisting autoimmune diseases).
7. Referral to a neurologist or center specializing in neuromuscular disease is advised for all patients with suspected MG and can be particularly important for complicated or refractory patients.
G. Transient neonatal myasthenia occurs in 10% to 15% of babies born to mothers with autoimmune MG. Within the first few days after delivery the baby has a weak cry or suck, appears floppy, and on occasion, requires mechanical ventilation. The condition is caused by maternal antibodies that cross the placenta late in pregnancy. As these maternal antibodies are replaced by the baby’s own antibodies, the symptoms gradually disappear, usually within a few weeks, and the baby is normal thereafter. Infants with severe weakness are treated with oral pyridostigmine 1 to 2 mg/kg every 4 hours.
H. Congenital myasthenia represents a group of rare hereditary disorders of the neuromuscular junction. The patients tend to have lifelong relatively stable symptoms of generalized fatigable weakness. These disorders are nonimmunologic, without ACh receptor antibodies, and therefore patients do not respond to immune therapy (steroids, thymectomy, and plasma exchange). Most of these patients improve on CEI. Even though there are many established subtypes of congenital MG, several are worth noting due in part to specific therapeutic implications. The fast channel congenital myasthenic syndrome tends to be static or slowly progressive, but usually very responsive to combination therapy with 3,4-diaminopyridine (enhances release of ACh) and pyridostigmine (reduces metabolism of ACh). Slow channel congenital myasthenic syndrome typically worsens over years as the endplate myopathy progresses. Although CEIs typically worsen symptoms, quinidine and fluoxetine, which reduce the duration of ACh receptor channel openings, are both effective treatments for slow channel syndrome. The congenital myasthenic syndrome associated with ACh receptor deficiency tends to be relatively nonprogressive and may even improve slightly as the patient ages. The disorder typically responds to symptomatic therapy with pyridostigmine and/or 3,4-diaminopyridine. Ephedrine produces benefit in some cases. Patients with endplate AChE deficiency usually present in infancy or early childhood with generalized weakness, underdevelopment of muscles, slowed pupillary responses to light, and either no response or worsening with CEIs. No effective long-term treatment has been described for congenital endplate AChE deficiency. A homozygous mutation of Dok-7 is responsible for a form of congenital myasthenia characterized by weakness in limbs and trunk but largely sparing the face, eyes, and oropharyngeal muscles. The formation of neuromuscular synapses requires the muscle-specific receptor tyrosine kinase (MuSK). Dok-7 is necessary and sufficient for the activation of MuSK. Albuterol was effective in treating patients with endplate acetylcholinesterase deficiency and also Dok-7 forms of congenital myasthenia.
LAMBERT–EATON MYASTHENIC SYNDROME
Lambert–Eaton myasthenic syndrome (LEMS) is a presynaptic disease characterized by chronic fluctuating weakness of proximal limb muscles. Symptoms include difficulty walking, climbing stairs, or rising from a chair. In LEMS there may be some improvement in power with sustained or repeated exercise. In contrast, eyelid ptosis, diplopia, dysphagia, and respiratory failure are far less common. In addition, LEMS patients often complain of myalgias, muscle stiffness of the back and legs, distal paresthesias, metallic taste, dry mouth, impotence, and other autonomic symptoms of muscarinic cholinergic insufficiency. LEMS is rare compared to MG, which is about 100 times more common. About half of LEMS patients have an underlying malignancy that is usually small-cell carcinoma of the lung. In patients without malignancy, LEMS is an autoimmune disease and can be associated with other autoimmune phenomena. In general, patients over age 40 are more likely to be men and have an associated malignancy whereas younger patients are more likely to be women and have no neoplasm malignancy. LEMS symptoms can precede detection of the malignancy by 1 to 2 years. Of all patients with small cell lung cancer, 4% have LEMS.
A. The examination typically shows proximal lower extremity weakness, although the objective bedside assessment may suggest relatively mild weakness relative to the patient’s history. The muscle stretch reflexes are absent. On testing sustained maximal grip there is a gradual increase in power over the initial 2 to 3 seconds (Lambert’s sign).
B. The diagnosis is confirmed with EMG studies, which typically show low amplitude of the compound muscle action potentials and a decrement to slow rates or repetitive stimulation. Following brief exercise, there is marked facilitation of the compound motor action potential (CMAP) amplitude. At high rates of repetitive stimulation, there may be an incremental response. SFEMG is markedly abnormal in virtually all patients with LEMS. The pathogenesis involves autoantibodies directed against voltage-gated calcium channels at cholinergic nerve terminals. These IgG antibodies also inhibit cholinergic synapses of the autonomic nervous system. Over 90% of LEMS patients demonstrated these antibodies to voltage-gated calcium channels in serum, providing another useful diagnostic test.
C. Treatment.
1. In patients with associated malignancy, successful treatment of the tumor can lead to improvement in the LEMS symptoms if the malignancy is successfully treated.
2. Symptomatic improvement in neuromuscular transmission may occur with the use of CEIs such as pyridostigmine.
3. 3,4-Diaminopyridine (DAP) increases ACh release by blocking voltage-dependent potassium conductance and thereby prolonging depolarization at the nerve terminal and enhancing the voltage-dependent calcium influx. 3,4-DAP has been shown to clearly improve most patients with LEMS with relatively mild toxicity and is becoming increasingly available, such that it represents first-line symptomatic therapy for LEMS. The typical beginning dose is 10 mg every 4 to 6 hours with gradual increase as needed up to a maximum of 100 mg/day. 3,4-DAP base and 3,4-DAP phosphate salt (amifampridine) two preparations.
4. Immunosuppressive therapy is used in patients with disabling symptoms. Long-term high-dose corticosteroids, plasma exchange, and IVIG have all been used with moderate success. In general, the use of these therapies should be tailored to the severity of patient’s symptoms.
BOTULISM
Consumption of sausage spoiled by Clostridium botulinum resulted in an outbreak of this paralytic illness in the 1700s in Germany, leading to the name botulism, derived from the Latin term for sausage, botulus. Botulinum toxin blocks ACh release at the presynaptic motor nerve terminal (and causes dysautonomia by blocking muscarinic autonomic cholinergic function as well). The intracellular target of botulinum toxin appears to be a protein of the ACh vesicle membrane. The toxin is a zinc-dependent protease that cleaves protein components of the neuroexocytosis apparatus.
A. Classic botulism occurs after ingestion of food contaminated by botulinum toxin. Eight different toxins have been identified, but disease in humans is caused by types A, B, and E. Type E is associated with contaminated seafood. All types produce a similar clinical picture, although type A may produce more severe and enduring symptoms. In all three types, the condition is potentially fatal. Most cases result from ingestion of bottled or canned foods that have not been properly sterilized during preparation, especially “home-canned foods.” Today’s tomatoes used in home canning may have a low acid content and therefore may be more vulnerable for contamination. Foods cooked on an outdoor grill and then wrapped in foil for a day or two, creating an anaerobic environment, can lead to toxin production. Home-bottled oils should also be considered; in the case of children, honey may be contaminated.
1. Clinical features begin 12 to 48 hours after ingestion of tainted food. Bulbar symptoms including diplopia, ptosis, blurred vision, dysarthria, and dysphagia occur initially, and are followed by weakness in the upper limbs and then in the lower limbs. In contrast to the typical patient with GBS, botulism is sometimes said to produce an acute “descending paralysis.” Severe cases result in respiratory failure, requiring mechanical ventilation. Botulism produces autonomic dysfunction, including constipation, ileus, dry mouth, and dilated pupils. (Note: Some of these signs are seen in most but not all patients; normal pupils do not rule out the diagnosis of botulism.)
2. Diagnosis. The CMAP amplitudes are typically low on the motor nerve conduction studies. Repetitive stimulation studies before and following exercise may show a decrement to low rates of repetitive stimulation and postexercise facilitation of the CMAP amplitude. It is wise to send both stool and serum specimens to the lab for detection of the toxin. The specimen is injected into the peritoneum of a mouse, while neutralized or inactivated specimen is injected as the control. If the mouse becomes paralyzed and dies, the diagnosis is secure. Toxin is found in blood samples 30% to 40% of the time, while stool samples have a somewhat higher yield (thus the need to send both). Newer polymerase chain reaction tests for the clostridial genes and ELISA identification of the toxin have been used to screen for the bacteria in food but are not widely available for clinical usage.
3. Management involves placement of the patient in the ICU and assiduous monitoring of pulmonary function every few hours. When the FVC falls below 15 mL/kg or below 1 L or if the patient appears to be having respiratory difficulty, intubation and mechanical ventilation are necessary.
a. There is a trivalent botulinum antitoxin, but its use is inconsistent, in part because of adverse side effects that occur in about 10% to 20% of patients. There is evidence that antitoxin shortens the course of the illness, especially the one associated with type E. If a diagnosis can be made early, it may be worth using the antitoxin. Serious complications from antitoxin therapy include serum sickness (4%), urticaria (3%), and anaphylaxis (2% to 3%).
b. The Center for Disease Control and Prevention (CDC) recommends administration of one vial of antitoxin for adult patients with botulism as soon as diagnosis is made, without waiting for laboratory confirmation; before administration of antitoxin, consider skin testing for sensitivity to serum or antitoxin. One vial of trivalent botulism antitoxin administered IV results in serum levels of type A, B, and E antibodies capable of neutralizing serum toxin concentrations in excess of those reported for botulism patients.
c. Antitoxin packages, including instructions for skin or conjunctival testing for hypersensitivity, are available through the CDC and state health departments. Antitoxin neutralizes toxin not yet bound to nerve terminals and has circulating half-life of 5 to 8 days. Patients who do not receive antitoxin treatment show free toxin in serum for up to 28 days.
4. Clinical course. With aggressive support, the overall mortality remains about 5% to 10%, usually the result of respiratory or septic complications. The other patients improve over a period of several weeks to several months. In those who survive, the eventual level of recovery usually is nearly complete. Several years after the illness, some patients have subjective fatigue and autonomic symptoms including constipation, impotence, and dry mouth. Clinical recovery results from brisk sprouting of new motor axons from the nerve terminal with reinnervation of denervated muscle fibers.
B. Infant botulism is probably the most frequent form of botulism. The infant ingests the spores of C. botulinum, which lodge in the intestinal tract, germinate there, and produce botulinum toxin in the gut. Honey has often been implicated as the contaminated food in the infant disease (25% of honey samples may contain spores). In adults, the small amount of C. botulinum in honey appears inadequate to colonize the GI tract. The typical presentation is an infant between the ages of 6 weeks and 6 months of age who exhibits generalized weakness and constipation. The weakness may start in the cranial muscles and then descend, causing a weak suck, a poor cry, and reduced spontaneous movement. The cranial muscles are weak, with poor extraocular movements, reduced gag reflex, and drooling. Finding C. botulinum in feces validates the diagnosis. The toxin is usually not detectable in the serum. EMG studies can point to the diagnosis in 80% to 90% of cases. Infantile botulism can range from mild to severe. Management centers on observation and general support (including respiratory stability). The recovery is usually excellent and runs a course of several weeks to several months. For infant botulism, IV botulinum immune globulin (BIG) trials in California were completed in early 1997 demonstrating safety and efficacy of human-derived BIG (BabyBIG) and a reduced mean hospital stay from 5.5 to 2.5 weeks. BIG is Food and Drug Administration (FDA) approved and is available from the California Department of Health Services. Antibiotic use is not recommended for infant botulism because cell death and lysis may result in the release of more toxin.
C. Wound botulism occurs when toxin is produced from C. botulinum infection of a wound. The symptoms are similar to those of classic botulism except that the onset may be delayed for up to 2 weeks after contamination of the wound. The diagnosis is supported by EMG studies, demonstration of toxin in the patient’s blood, or finding the organism in the patient’s wound. Wounds that lead to botulism include direct trauma, surgical wounds, and wounds associated with drug use (such as IV and intranasal cocaine). The use of local antibiotics such as penicillin G or metronidazole may be helpful in eradicating C. botulinum in wound botulism.
Key Points
• Half of all MG patients present with either diplopia or ptosis or both and within several months 80% of patients have ocular involvement.
• 20% of MG patients have pure ocular disease, 20% have a spontaneous long-lasting remission, and 10% have thymoma.
• A recent prospective controlled trial has demonstrated clear benefit of thymectomy in the treatment of MG.
• Patients with MuSK myasthenia who are refractory to conventional first-line treatment respond disproportionately well to plasma exchange and rituximab.
• Patients with Lambert–Eaton syndrome present with fluctuating proximal lower extremity weakness, dry mouth, and absent muscle stretch reflexes and half of them have small cell lung cancer.
• The most effective first-line treatment for Lambert–Eaton syndrome is 3,4 diaminopyridine.
Related posts:
 Approach to the Patient with Acute Confusional State (Delirium/ Encephalopathy)
Approach to the Patient with Sleep Disorders
Approach to the Patient with Hearing Loss
Approach to the Patient with Lower Extremity Pain, Paresthesias, and Entrapment Neuropathies
Approach to Common Office Problems of Pediatric Neurology
Movement Disorders
Approach to the Patient with Acute Confusional State (Delirium/ Encephalopathy)
Approach to the Patient with Sleep Disorders
Approach to the Patient with Hearing Loss
Approach to the Patient with Lower Extremity Pain, Paresthesias, and Entrapment Neuropathies
Approach to Common Office Problems of Pediatric Neurology
Movement Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


