Abstract
Administration of omega 3 polyunsaturated fatty acids (n-3 PUFAs) is a biologically plausible, yet clinically untested, candidate therapeutic strategy to prevent or blunt neurologic disability after traumatic brain injury (TBI). The biologic plausibility of n-3 PUFAs rests on evidence that supports at least three putative mechanisms of neuroprotection after TBI. First, n-3 PUFA supplementation may address an acquired or existing n-3 PUFA deficiency which, if present, could impair recovery from TBI. Second, enzymatic and nonenzymatic metabolism of n-3 PUFAs may yield derivatives that modulate neuroinflammation after TBI. Third, n-3 PUFA supplementation may blunt oxidative injury after TBI via a free-radical scavenging mechanism. This chapter reviews the current state of bench and clinical research in this area and identifies potential future avenues of investigation.
Keywords
Inflammation, Lipidomics, Membrane characteristics, Oxidative stress, PUFAs
Docosahexaenoic Acid and Omega 3 Fatty Acids
Administration of omega 3 polyunsaturated fatty acids (n-3 PUFAs) is a biologically plausible, yet clinically untested, candidate therapeutic strategy to prevent or blunt neurologic disability after traumatic brain injury (TBI). The biologic plausibility of n-3 PUFAs rests on evidence that supports at least three putative mechanisms of neuroprotection after TBI. First, n-3 PUFA supplementation may address an acquired or existing n-3 PUFA deficiency which, if present, could impair recovery from TBI. Second, enzymatic and nonenzymatic metabolism of n-3 PUFAs may yield derivatives that modulate neuroinflammation after TBI. Third, n-3 PUFA supplementation may blunt oxidative injury after TBI via a free-radical scavenging mechanism. This chapter will review the current state of bench and clinical research in this area and will identify potential future avenues of investigation.
Omega 6 and Omega 3 Polyunsaturated Fatty Acids: A Brief Overview
PUFAs are lipids with a hydrocarbon tail that is between 18 and 22 carbons long and includes at least one double bond. Omega 3 and omega 6 PUFAs are characterized by having a double bond located three and six carbons away, respectively, from the “omega” carbon or the methyl end of the hydrocarbon chain ( ).
Mammals must obtain the short-chain PUFAs alpha linolenic acid (ALA, 18:3n-3) and linoleic acid (LA, 18:2n-6) from the diet because they cannot perform de novo synthesis of these fatty acids. ALA and LA are depicted schematically in Fig. 18.1 . ALA and LA are commonly found in plant-based foods such as nuts, seeds, and their derived oils.

Conversion of ALA and LA to the respective n-3 and n-6 long chain PUFAs via elongation and desaturation occurs in a limited fashion, modulated by sex and the dietary ratio of ALA to LA, among other factors ( ). Long-chain n-3 and n-6 PUFAs are important components of cell membrane phospholipids, particularly in neural tissue where arachidonic acid (AA, 20:4n-6) and docosahexaenoic acid (DHA, 22:6n-3) predominate. Eicosapentaenoic acid (EPA, 20:5n-3) is another important neural PUFA, albeit present at concentrations about 250-fold lower than DHA ( ). AA, DHA, and EPA are depicted schematically in Fig. 18.2 .

Phospholipids are characterized by a glycerol backbone with a polar head to which two fatty acids are attached, usually via ester linkages. Typically, a saturated fatty acid is attached to the sn-1 position of the glycerol backbone, while omega 3 or omega 6 PUFAs are attached to the sn-2 position ( ).
Very little AA exists in the diet. Thus, the vast majority of AA derives from ingested LA. In contrast, since DHA and EPA are very inefficiently synthesized from ALA, they are generally derived from dietary sources such as salmon, tuna, other fatty fish, and algae. Only small amounts of EPA are found in neural cell membranes because dietary EPA is either metabolized or used as a DHA precursor. Thus, most of the DHA content of the mammalian central nervous system reflects dietary intake of DHA and EPA ( ). Immature as well as gestating mammals require higher dietary intake of DHA and DHA precursors than do adults, to account for the rapid DHA accretion of the developing brain. In utero, most DHA accretion occurs in the latter part of pregnancy while the rate of postnatal DHA accretion parallels brain development ( ).
Brain DHA Content is Highly Conserved
Brain DHA is found in cellular and organellar, including mitochondrial, membranes ( ). The vast majority of brain DHA is esterified in phospholipids, predominantly in a type of ether phospholipid, ethanolamine-containing plasmalogens ( ). Brain DHA is highly conserved, even in the face of dietary inadequacy that produces DHA deficiency in nonneural tissues. Experimental induction of brain DHA deficiency requires exposure to prolonged periods of dietary insufficiency because DHA consumption in the brain is limited to the small amounts lost to energy generation and to oxidative damage. Damaged DHA is deesterified from its sn-2 position by type VI calcium-independent phospholipases A2 (iPLA2 beta and gamma) or, possibly, by a plasmalogen-specific PLA2. Such free DHA may be metabolized enzymatically or nonenzymatically into biologically active compounds or it may be reintegrated into membranes. In health, only small amounts of free DHA are metabolized by lipoxygenases (LOX), cyclooxygenases (COX), and other enzymes; instead, released DHA is usually reincorporated into membrane phospholipids by remodeling processes that are not yet well understood, as outlined below ( ).
Free DHA may be reincorporated into membrane phospholipids by acetylation to coenzyme A (CoA) by an acyl-CoA synthetase (ACSL), an ATP-consuming process, followed by the action of an acyl-CoA:lysophospholipid acyltransferase (LPLAT). ACSL6 is a candidate ACSL given its enrichment in the brain and its specificity for DHA. While a type of LPLAT (LPEAT2) is highly expressed in the brain and has selective action on ethanolamine plasmalogens, the DHA specificity of LPEAT2 and other brain-enriched LPLATs is unknown ( ). In summary, though the details of brain DHA accretion are under active investigation, it is abundantly clear that brain DHA content is highly conserved in health.
Role of Omega-3 PUFAs in Normal Brain Development and Function
DHA is the major n-3 PUFA esterified in retinal and brain cell membranes. Brain and retinal DHA content increases dramatically during early development, suggesting that DHA is important for normal neurologic function.
Animal studies have shown that decreases brain and retinal DHA content resulting from dietary DHA deficiency during development is associated with impairments in membrane fluidity, synaptic function, and neurotransmitter release and with decreased visual, cognitive, and behavioral performance ( ). DHA-deficient brains display a marked increase in the AA-derived DPA 5n-6, a change that would hypothetically decrease membrane flexibility and affect the functionality of integral membrane proteins ( ). DHA deficiency also decreases synaptic connectivity and the length and number of dendritic branches ( ).
In preterm infants fed a commercial, non-DHA supplemented formula, low red blood cell and plasma DHA levels correlated with decreased visual acuity as measured by visual-evoked responses at both 36 and 57 weeks postconceptional age, compared to preterm infants fed human milk or n-3 PUFA-fortified commercial formulas ( ). In adult humans, epidemiological and intervention studies have linked lower DHA intake and/or lower DHA levels in plasma and blood cell lipids to an increased risk of dementia and cognitive decline ( ).
In contrast to studies of DHA deficiency, clinical studies of n-3 PUFA supplementation have yielded mixed results. Possible explanations for such variability include inconsistent study design, small study sizes, and the inability to accurately assess baseline DHA sufficiency. Some studies of n-3 PUFA supplementation during pregnancy and/or infancy showed no effect, while others found that the n-3 PUFA-supplemented groups displayed improvements in specific neurodevelopmental outcomes such as visual-evoked potentials and neurodevelopmental scores ( ). Studies on the cognitive effects of DHA and/or EPA supplementation in school-aged children and adults are similarly inconclusive. Supplementation generally appears to improve cognitive performance, though effects are more often noted in those individuals with a lower baseline performance and/or poor DHA intake ( ). For example, a double-blind, randomized, placebo-controlled trial (RCT) in healthy children aged 7–9 showed that DHA supplementation improved reading performance limited to those children at the lowest reading percentile at baseline ( ) and a meta-analysis of interventional trials revealed that DHA improved episodic memory in those adults with mild memory complaints ( ).
Role of DHA in Normal Neural Membrane Composition and Function
The phospholipid bilayer, composed of saturated and unsaturated fatty acids, is the backbone of all cellular, synaptosomal, and vesicular membranes. The length of these fatty acids and the number of double bonds they contain determine how tightly the hydrocarbon tails may be packed together and thus the fluidity and thickness of the cell membrane. DHA, with its 22 carbons and 6 double bonds, is a very long and highly unsaturated PUFA. Higher DHA content promotes greater fluidity, thinness, compressibility, permeability, and a higher fusion rate of cell membranes ( ). DHA membrane content is very high in the mammalian brain, particularly in synaptosomes where DHA content is as high as 35% of all fatty acids ( ). Lipid composition of brain membranes affects protein-related processes via a number of mechanisms. Lipid composition helps determine the type and amount of integral proteins present and the ability of membrane proteins to translocate/rotate appropriately. Membrane lipids can provide enzyme cofactors and can be modified to act as second messengers in intra- or extracellular signaling such as neurotransmitter release. DHA deficiency has been linked to specific anomalies in many of these processes ( ).
A DHA-rich diet promotes higher brain DHA content and greater membrane fluidity in health and disease. Healthy mice fed diets rich in n-3 PUFAs had higher brain phospholipid DHA levels, greater synaptic membrane fluidity, and improved learning compared to those fed a diet high in saturated fats ( ). In a mouse model of Alzheimer’s disease, oral DHA supplementation abrogated the cognitive dysfunction, and the decreased synaptosomal membrane fluidity induced by the model ( ). Finally, in adult rats, 7 days of oral fish oil normalized the TBI-induced deficits in striatal evoked dopamine release, suggesting that DHA improved vesicular or synaptic function after TBI ( ).
Preexisting DHA Deficiency Worsens Outcomes After Experimental TBI
A preexisting DHA deficiency is associated with worse outcomes after experimental TBI. Feeding mice a diet free of n-3 PUFAs over three generations reduced adult mouse brain membrane DHA content by 70% with a corresponding increase in the content of AA and AA derivatives, compared to mice on a regular diet. Relative to mice on the regular diet, the DHA deficient mice had worse functional and histologic outcomes after experimental TBI ( ). Similarly, in juvenile rats exposed to dietary manipulations to decrease brain DHA content before experimental TBI, a greater brain DHA deficit correlated with worse functional deficits after injury ( ). Adverse effects of a preexisting DHA deficiency on TBI-related outcomes could result from suboptimal function of those neural membranes re-formed in the absence of adequate dietary DHA/DHA precursor intake. Alternatively, or in addition, enzymatic transformation of free AA rather than free DHA, to produce a predominance of proinflammatory lipid mediators (see Fig. 18.3 ) may adversely affect outcomes.
Possible Role of an Acquired DHA Deficiency After TBI
If TBI increased the rate of brain DHA consumption to exceed its rate of accretion, then TBI could produce an acquired DHA deficiency with negative effects on neural membrane composition as described in models of brain DHA deficiency. Brain DHA losses after TBI could result from increased enzymatic and nonenzymatic metabolism of deesterified DHA and/or the failure to reincorporate deesterified DHA into neural membranes. Failure of reincorporation could occur if ATP availability or functionality of this membrane remodeling pathway decreased after TBI. In that setting, the typical western diet with its low dietary DHA intake would only further aggravate the deficit. Indeed, the phospholipid DHA to AA ratio and total PUFA content decreased in hippocampi and cortices of injured mice 3 months after controlled cortical impact (CCI) ( ). In rats after fluid percussion, experimental TBI decreased DHA content of rat brain cortices 2 weeks after injury ( ). Similarly, we have preliminary data that rat brain DHA content 2 months after injury in rats exposed to CCI at day 17 of life decreased relative to sham-injured controls (unpublished data). It is unknown whether or not the human brain experiences loss of DHA content after TBI. Future studies may shed light on this question, possibly by using positron-emission tomography and other in vivo methods to measure brain DHA content ( ).
Deesterification of fatty acids from membrane phospholipids is mediated by activated PLA2 enzymes. There are four classes of PLA2 enzymes, each with many isoforms having variable fatty acid specificity, regional expression, and activation properties. Depending on the type of PLA2 activation, different deesterified fatty acids including DHA and AA would be released for enzymatic and/or nonenzymatic metabolism or for potential reintegration into membrane phospholipids ( ).
TBI increases cytoplasmic and plasmalogen-specific (cPLA2 and plsPLA2) activity after TBI ( ). At the time of this writing, there are no published studies on the effects of TBI on the activity of the DHA-specific iPLA2 enzyme. However, it is quite likely that TBI affects it, since i-PLA2 is responsible for more than 70% of whole-brain PLA2 activity and for most of PLA2 activity in brain mitochondria. In addition, iPLA2 is present in microglia, an important component of the brain’s cellular response to TBI ( ). In health, iPLA2 remodels the mitochondrial membrane by removing oxidized fatty acids from mitochondrial membrane lipids. In ATP-depleted conditions, however, released fatty acids may be metabolized instead of being reintegrated into membranes. CCI activated phospholipases A2 and C and increased free DHA and AA levels in the damaged rat cortex, primarily AA at 30 min and DHA at 24 h after injury ( ).
Clinical studies support the contention that free fatty acid losses occur after TBI. In a study of eight TBI patients, Farias et al. showed that DHA and AA levels in CSF increased relative to controls during the first 4 days after injury, with highest levels noted on the first day ( ). In another clinical study, TBI also increased levels of free DHA and other fatty acids in CSF. In this study, greater elevations correlated with poor outcome after TBI ( ).
In summary, a preexisting DHA deficiency worsens outcomes after experimental TBI. TBI may induce an acquired DHA deficiency in the brain if deesterified DHA after TBI is lost and inadequately replaced. In this context, at least a part of the neuroprotective effects of DHA supplementation after TBI could be ascribed to prevention of an acquired DHA deficiency. On the other hand, as outlined in the next section, the predominance of DHA rather than AA as a substrate for enzymatic metabolism after TBI may be neuroprotective via an inflammation-resolving pathway. Further study is needed to elucidate the relative importance of these putative neuroprotective mechanisms. Results of such research would inform dosing paradigms because dose dependency would be expected for DHA-associated neuroinflammatory effects while the dose of DHA needed merely to correct a deficiency should be lower and manifest a ceiling effect ( )
Effects of n-3 PUFAs on Neuroinflammation
Administration of n-3 PUFAs may improve neurologic outcomes by modulating the inflammatory response after TBI. Inflammation is an injury response characterized by increased vascular permeability, altered vascular tone, and the influx of granulocytes and other immune cells to the afflicted area. Such a response, if swift and effective, is often beneficial. On the other hand, failure to resolve inflammation is associated with numerous diseases, including neurodegeneration, and with worse outcomes after TBI ( ). Resolution of inflammation is characterized by tissue repair, regeneration, and reversal of leukocyte invasion by mechanisms such as apoptosis and phagocytosis ( ). Neuroinflammation is largely mediated by astrocytes, microglia (the brain’s resident macrophages), and infiltrating macrophages. n-3 PUFAs are thought to exert effects on neuroinflammation via enzymatic and nonenzymatic pathways and via effects on gene transcription.
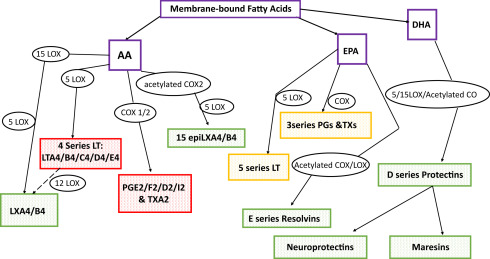
Enzymatic metabolism of fatty acids deesterified and thus liberated from neural membranes by activated PLA2 may produce proinflammatory, antiinflammatory, and inflammation-resolving mediators, as shown in Fig. 18.3 ( ). Eicosanoids are signaling molecules derived from 20 carbon free fatty acids, primarily from AA. A specific eicosanoid may mediate responses such as vasodilation, platelet aggregability, vascular permeability, or leukocyte chemotaxis.
Enzymatic modification of AA generally produces proinflammatory molecules such as the 4 Series Leukotrienes (LTA4, B4, C4, D4, and E4), the 2 Series Prostaglandins (PGE2, F2, D2, I2), and the 2 Series Thromboxane A2 (TXA2) resulting from LOX and COX activities, respectively. Some AA products are antiinflammatory and proresolving mediators, such as the lipoxanes LXA4 and B4 and the 15 epi-lipoxanes (15epi-LXA4/B4). Indeed, blocking AA conversion to LTs using certain LOX inhibitors before or after TBI offers beneficial effects in adult rodents ( ).
Enzymatic modification of EPA and DHA generally produces antiinflammatory and proresolving molecules. Increasing the EPA and DHA content of the diet will shift eicosanoid production from AA toward EPA, commonly resulting in a reparatory or a less proinflammatory state ( ). Cyclooxygenase 2 (COX2), in the presence of aspirin, will produce D and E series resolvins from DHA and EPA, respectively. Resolvins have potent antiinflammatory and inflammation-resolving activities ( ). Other DHA derivatives include the nonaspirin-dependent D-resolvins, produced via the action of 15-LOX, and the protectins. Neuroprotectin D1 (NPD1) is a protectin made in neural tissues that results from 15-LOX activity followed by epoxidation and hydrolysis reactions. NPD1 has antiapoptotic and antiinflammatory effects on various cell-signaling cascades in the brain ( ). Blockade of resolvin or protectin production via COX2 or LOX inhibitors results in prolonged inflammation secondary to impaired phagocytic removal and repair processes ( ). Generally speaking, then, AA derivatives are proinflammatory while DHA/EPA derivatives are antiinflammatory and/or inflammation resolving.
Nonenzymatic lipid derivatives primarily result from free-radical attack on membrane-bound PUFAs, followed by hydrolysis and release of free oxidized PUFAs into the tissues and circulation. These oxidized PUFAs are useful biomarkers of oxidative stress. Recently, it has become clear that they are also biologically active molecules. For example, peroxidation products of DHA and EPA decrease leukocyte adhesion and proinflammatory cytokine production ( ).
Recent evidence suggests that many potent in vivo effects of n-3 PUFAs on inflammation are mediated by gene transcription. In vitro and in vivo, DHA and EPA decrease mRNA and protein levels of the inflammatory cytokines tumor necrosis factor alpha (TNFα), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), among others. DHA and EPA decrease activation of the transcription factor nuclear factor κ B (NF κ B), possibly by altering membrane properties and thus lipid raft formation, or via direct activation of the fatty acid receptor protein peroxisome proliferator-activated receptor gamma (PPAR γ) ( ).
Mouse experiments support the concept that a shift toward greater n-3/n-6 PUFA ratio in neural tissues decreases neuroinflammation. Relative to wild-type mice, transgenic “FAT-1” mice rendered n-3 PUFA-rich by insertion of a C . elegans PUFA desaturase that converts n-6 PUFAs to n-3 PUFAs, respond to intraperitoneal (IP) lipopolysaccharide injection with a blunted inflammatory cytokine response and with protection from the expected inflammation-induced cognitive impairments ( ).
Related posts:
 Hypothermia for Traumatic Brain Injury: Current Evidence and Future Directions
The Future of TBI: Hyperbaric Oxygen as a Primary Therapeutic Approach
Erythropoietin and Its Derivatives : Mechanisms of Neuroprotection and Challenges in Clinical Translation
Voltage-Gated Calcium Channel Blockers for the Treatment of Traumatic Brain Injury
5-Lipoxygenase-Activating Protein Inhibitors: Promising Drugs for Treating Acute and Chronic Neuroinflammation Following Brain Injury
Memantine : A Safe and Tolerable NMDA Antagonist with Potential Benefits in Traumatic Brain Injury
Hypothermia for Traumatic Brain Injury: Current Evidence and Future Directions
The Future of TBI: Hyperbaric Oxygen as a Primary Therapeutic Approach
Erythropoietin and Its Derivatives : Mechanisms of Neuroprotection and Challenges in Clinical Translation
Voltage-Gated Calcium Channel Blockers for the Treatment of Traumatic Brain Injury
5-Lipoxygenase-Activating Protein Inhibitors: Promising Drugs for Treating Acute and Chronic Neuroinflammation Following Brain Injury
Memantine : A Safe and Tolerable NMDA Antagonist with Potential Benefits in Traumatic Brain Injury
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


