EEG of Degenerative Disorders of the Central Nervous System
John Gaitanis
INTRODUCTION
Degenerative disorders of the central nervous system encompass a wide range of genetic and metabolic conditions, which result in progressive neurologic disability. Senile dementia can be included under this heading, but is discussed elsewhere in the text. Metabolic encephalopathies are distinct from chronic, degenerative conditions in that they often result from systemic disorders and are commonly reversible. Given the wide array of etiologies for degenerative diseases, they can be categorized in multiple ways. The most common classification system is to separate these disorders based on the type of cellular organelle that is primarily involved, as is done in this chapter. Special attention is also given to the predominant neuroanatomical structures involved, since this allows for an EEG to neuroanatomical correlation (Table 15-1).
LYSOSOMAL DISORDERS
Lysosomes are membrane-bound organelles that function as the digestive system of the cell. They contain enzymes that break down macromolecules originating either internally or externally. Lysosomal storage disorders occur when there is deficiency of one or more of these enzymes. Undegraded material accumulates, leading to cellular and organ dysfunction. Most of these conditions exhibit autosomal recessive inheritance.
Table 15.1 | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
GM2 Gangliosidoses/Tay-Sachs Disease
The infantile form of Tay-Sachs disease is named for the British ophthalmologist Warren Tay, who, in 1881, described the cherry-red spot, and the American neurologist Bernard Sachs, who reported on the clinical features and the enlarged pyramidal neurons of the condition. The biochemical understanding came about when Ernst Klenk found a novel group of glycolipids in the brains of these patients, which was later discovered to be GM2 ganglioside. Its accumulation in neurons results from a deficiency of the lysosomal enzyme hexosaminidase A. The hexosaminidase enzyme is comprised of alpha and beta subunits. Hexosaminidase A consists of one alpha and one beta subunit and hexosaminidase B is composed of two beta subunits. In Tay-Sachs disease, there is loss of the alpha subunit resulting in absence of hexosaminidase A.
The result is a neurologically devastating disease that begins in the first months of life. The initial symptom is excessive startle myoclonus to noise, sound, or touch. The startle does not attenuate with subsequent exposures, distinguishing it from a normal Moro response. The myoclonic movements consist of arm and leg extension. As the disease progresses, motor, visual, and intellectual impairments develop. The affected infant develops axial hypotonia with appendicular spasticity and increased reflexes. Motor skills, such as head control, rolling, and purposeful hand movements, are lost. Visual loss ensues as lipid accumulates in the retinal ganglion cells, resulting in a whitish discoloration around the fovea—the characteristic cherry-red spot. With intraneuronal accumulations of GM2 ganglioside, macrocephaly develops. The children experience progressive cognitive impairment and seizures. Feeding and autonomic disturbances develop. Between 3 and 5 years of age, the patient succumbs to cachexia or aspiration pneumonia.
The disease affects multiple regions of the brain. The basal ganglia and cerebral white matter show low-density signal on CT in early stages and enlargement of the caudate nucleus later in the disease course (1). Early into the disease, the interictal EEG may be normal. By age 1, there is a rapid and progressive deterioration of the EEG (2). Paroxysmal features are not prominent, but during myoclonic seizures, widespread spike and sharp waves are seen (3). A progressive decline in voltage occurs in later stages of the disease, likely resulting from continued
neuronal loss. The electroretinogram remains normal even in advanced stages (2,4), since only the ganglion cell layer of the retina is affected. Progressive loss of the VEP occurs between 9 and 15 months (2).
neuronal loss. The electroretinogram remains normal even in advanced stages (2,4), since only the ganglion cell layer of the retina is affected. Progressive loss of the VEP occurs between 9 and 15 months (2).
Sandhoff disease, resulting from mutations of the beta subunit of hexosaminidase A, is phenotypically similar to Tay-Sachs disease, the only differences being the presence of hepatosplenomegaly, cardiomyopathy, or N-acetylglucosamine-containing oligosaccharides in the urine of patients with Sandhoff disease. The AB variant occurs when there is a deficiency of the GM2 activator, which is necessary for hydrolysis of GM2 gangliosides by hexosaminidase A. Its phenotype is indistinguishable from the infantile form of Tay-Sachs (5). Late-onset variants of Tay-Sachs also exist, and can manifest in childhood, adolescence, or adulthood. They may result in a wide constellation of symptoms including spastic paraparesis, ataxia with cerebellar atrophy, seizures, or psychiatric symptoms. Psychosis and depression can even be the initial manifestation in adult patients (6). EEG changes in late-onset GM2 gangliosidosis are variable and unrelated to age or enzyme defect (2).
GM1 Gangliosidosis
GM1 gangliosidosis results from a deficiency of β-galactosidase, which is responsible for cleavage of the terminal galactose of GM1. The result is neuronal storage of monosialoganglioside GM1. Histopathology reveals a marked decrease in the number of oligodendrocytes and myelin sheaths. As a result, spasticity, tonic spasms, and pyramidal signs predominate. As in Tay-Sachs disease, a cherry-red spot is seen and the ERG remains normal (2). The EEG also shows a progressive deterioration as it does in GM2 patients. EEGs universally reveal abnormal slowing, which worsens as the disease advances. By 2 to 3 years of age, 4 to 5 cycle/sec rhythmic activity is seen in the temporal regions. Paroxysmal discharges, however, are not commonly observed in GM1 patients (7).
Neuronal Ceroid Lipofuscinoses
The neuronal ceroid lipofuscinoses (NCL) are a group of neurodegenerative conditions caused by intralysosomal accumulation of ceroid and lipofuscin (8). Originally described as a form of “amaurotic familial idiocy,” the group of disorders was renamed NCL by Zeman to better distinguish them from gangliosidoses (9). Clinically, the disease group is characterized by cognitive decline, progressive myoclonic epilepsy, and motor regression. Some forms also cause visual failure (8). The four originally described phenotypes are organized according to age of onset: infantile (INCL), late infantile (LINCL), juvenile (JNCL), and adult neuronal ceroid lipofuscinosis (ANCL). Patients with INCL are normal at birth, and exhibit developmental delays or myoclonic seizures between 6 and 24 months of age. In LINCL, patients experience developmental regression and epilepsy between 2 and 4 years. JNCL develops between 4 and 10 years with visual loss being the initial symptom and epilepsy following shortly afterwards. ANCL appears sometime around 30 years of age and presents with progressive myoclonic epilepsy, dementia, behavioral abnormalities, ataxia, and pyramidal or extrapyramidal signs (10). These disorders are inherited in an autosomal recessive pattern and there are at least seven genes responsible for the clinical phenotypes.
MRI findings in INCL include cerebral atrophy, callosal thinning, T2-weighted hyperintensities in the thalami, and cerebellar atrophy. After 4 years of age, the atrophy is severe and the entire white matter shows abnormally high signal intensity. SPECT studies show progressive cerebral and cerebellar hypoperfusion with relative sparing of the basal ganglia. LINCL and JNCL reveal lesser degrees of atrophy (11).
The EEG findings reflect the predominance of cortical abnormalities on MRI and SPECT. In INCL, the EEG initially shows a lack of attenuation to eye opening and a disappearance of sleep spindles. The background activity gradually attenuates and becomes flat by 3 years of age. ERGs are unrecordable by age 3 and VEPs are unrecordable by 4 years. There is progressive attenuation of cortical somatosensory potentials (12). In LINCL, seizures are the presenting symptom, and the EEG reveals occipital spike waves in response to photic stimulation at 1 to 2 Hz. The ERG is abnormal at presentation and extinguishes in time. VEPs are enhanced, but diminish in the final stages of the disease (13). In JNCL, the EEG shows disorganization and spike- and slow-wave complexes. ERG and VEPs are abnormal early into the course of the disease. The EEG abnormalities of ANCL are nonspecific (14). In one case series, the frequency of background slowing in all types of NCL was 94.6% and epileptiform discharges were seen in 81.1% (15). Slow-frequency photic stimulation elicited a response in 5 out of 22 patients, a giant SSEP was seen in 7 of 25 patients, and slow or absent waveforms were seen in 9 of 25 patients on VEP (15).
Mucopolysaccharidoses
The mucopolysaccharidoses (MPSs) are a heterogeneous group of disorders caused by deficiency of a lysosomal enzyme involved in the degradation of glycosaminoglycans (16). As glycosaminoglycans accumulate in lysosomes, cell and organ dysfunction result. Seven types exist. The age of onset and severity vary, but most exhibit a chronic, progressive course characterized by multisystemic involvement, coarse facies, organomegaly, and bony defects (dysostosis multiplex) (16). The more severe forms (MPS I, MPS II, and MPS VIII) result in progressive mental retardation, whereas patients with MPS IV and MPS VI have normal cognition, even when somatic manifestations are severe (16). The only variant without somatic symptoms is MPS III, of which there are four subtypes. MPS III presents between 1 and 6 years of age with delayed psychomotor development and behavioral problems. Progressive dementia, sleeplessness, and epilepsy follow (17). In MPS III, the EEG is normal in half of patients, but shows diffuse slowing in the rest (18). Low-amplitude 12- to 15-Hz activity is seen intermixed with generalized delta frequencies (19). These findings correspond to the severe cortical involvement seen pathologically (19). VEPs and BAEPs are almost always normal (18). Enzyme replacement therapy and hematopoietic stem cell transplantation offer potential benefit in these conditions (20,21).
Sialidosis Types I and II (Neuraminidase Deficiency)
In sialidosis, a deficiency of the enzyme neuraminidase leads to progressive storage of sialidated glycopeptides and oligosaccharides. Sialidosis bears clinical and histopathologic resemblance to the mucopolysaccharidoses, in that patients exhibit mild Hurler-like facies, skeletal dysplasia, and psychomotor retardation. Sialidosis is divided into types I and II (22). Type I has a later age of onset and patients exhibit little if any dysmorphology. Visual failure, cherry-red spots, ataxia, and myoclonus are common features (23,24). Type II patients have an earlier age of onset and demonstrate coarse facies, hepatosplenomegaly, and skeletal anomalies (dysostosis multiplex and spinal deformities) (25). MRI findings include atrophy of the cerebral hemispheres, cerebellum, and pontine region (26).
EEG findings in sialidosis types I and II are indistinguishable (27). In type I, the background is low voltage and rhythmic spikes are observed over the vertex, which increase in frequency during sleep (27). Spike-wave discharges precede myoclonic jerking in both types I and II (28,29). VEPs demonstrate low amplitudes and prolonged latencies in type I (28), while SEPs exhibit giant potentials in both conditions (28,29).
Niemann-Pick Disease
Niemann-Pick disease occurs when a deficiency of the enzyme acid sphingomyelinase (ASM) causes accumulation of sphingomyelin within cells of the monocyte-macrophage system (30). Four phenotypic subtypes exist. In Niemann-Pick disease type A (NPD-A), infants present in the first few weeks or months of life with failure to thrive and hepatomegaly. Psychomotor regression follows, with head lag, inability to sit, and loss of interest in surroundings. In time, rigidity and opisthotonus predominate. Death occurs in the second or third year. Startle myoclonus, like that seen in Tay-Sachs, is not observed and there are no coarse features, like those present in mucopolysaccharidosis. In Niemann-Pick type B (NPD-B), visceral involvement predominates without significant neurologic impairment (31). An intermediate form between NPD-A and NPD-B may also exist with retinal degeneration and slowed nerve conduction presenting in adolescence or adulthood (30). In Niemann-Pick type C (NPD-C), patients present between ages 3 and 8 years with ataxia, vertical supranuclear gaze palsy, and hepatosplenomegaly. In time, they experience dementia, dystonia, and seizures (32). Cataplexy occurs late in the disease course (33). Niemann-Pick type D (NPD-D) occurs in French Canadian patients who share a common founder mutation (34).
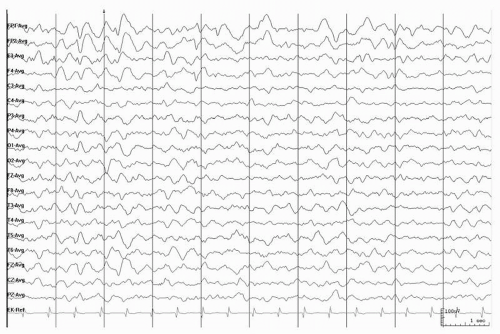 Figure 15.1 EEG in a 10-year-old boy with Niemann-Pick type C. Note the frontal slowing with intermixed sharp waves bilaterally. |
In late childhood or adult-onset NPD-C, the EEG may reveal generalized slowing (Fig. 15.1) (35). Late into the course of NPD-C, abnormally high-voltage and diffuse alpha activity is seen and is enhanced by intermittent photic stimulation. In a patient with myoclonus, EEG-EMG frequency analysis demonstrated a cortical origin for the myoclonus (36).
Gaucher Disease
Gaucher disease is the most common lysosomal storage disorder. It results from a deficiency of the lysosomal enzyme beta-glucocerebrosidase, resulting in accumulation of
glucocerebroside in the mononuclear phagocyte system (37). The characteristic features include hepatosplenomegaly, bony disease, and neurologic decline. Three phenotypic subtypes exist. Type I has minimal if any neurologic impairment. Bulbar symptoms predominate in type II patients (38). In type III patients, ocular motor apraxia is often the initial finding. The pathologic hallmark of the disorder is the Gaucher cell, which is an enlarged macrophage with cytoplasmic linear inclusions resulting from excessive lysosomal accumulation of glucocerebroside (38). Enzyme replacement therapy with recombinant glucocerebrosidase (Cerazyme) is the mainstay of treatment in Gaucher disease (39), but is not effective for the neurologic symptoms since it does not cross the blood-brain barrier.
glucocerebroside in the mononuclear phagocyte system (37). The characteristic features include hepatosplenomegaly, bony disease, and neurologic decline. Three phenotypic subtypes exist. Type I has minimal if any neurologic impairment. Bulbar symptoms predominate in type II patients (38). In type III patients, ocular motor apraxia is often the initial finding. The pathologic hallmark of the disorder is the Gaucher cell, which is an enlarged macrophage with cytoplasmic linear inclusions resulting from excessive lysosomal accumulation of glucocerebroside (38). Enzyme replacement therapy with recombinant glucocerebrosidase (Cerazyme) is the mainstay of treatment in Gaucher disease (39), but is not effective for the neurologic symptoms since it does not cross the blood-brain barrier.
MRI in type II disease is often normal, but cerebral atrophy is commonly observed in type III patients (40). BAERs and SSEPs can demonstrate abnormalities, even when no lesions are detectable by MRI (41). EEGs show rhythmical sharp waves (6 to 10/sec), polyspikes, and spike-wave discharges in type III patients (42). These features are diffuse, but have a posterior predominance. EEGs are commonly normal in type II patients. Reduced amplitude can also be an EEG feature of some patients (41).
Fabry Disease
Fabry disease is the only X-linked lysosomal disorder; the others are inherited in an autosomal recessive fashion. Hence, the disease affects men more severely than it does women (43). It results from a deficiency of alpha-galactosidase A. This leads to storage of glycosphingolipids in a variety of tissues, producing cellular dysfunction, inflammation, and fibrosis. Symptoms begin in early childhood with peripheral neuropathy and burning pain of the hands and feet, which can remain disabling throughout life (43). Hypohydrosis, nausea, and postprandial diarrhea are also seen in childhood. By adulthood, proteinuria and renal failure develop. Left ventricular hypertrophy and coronary artery disease are also common. Angiokeratoma develop on the skin. Neurologically, cryptogenic strokes occur. Enzyme replacement therapy can clear microvascular endothelial deposits from the kidneys, heart, and skin, reversing the pathogenesis (44). MRI scans reveal gray and white matter lesions. The anterior circulation generally appears normal, but tortuous basilar arteries are common (45). EEGs can reveal either focal or diffuse slowing (46).
Metachromatic Leukodystrophy
Metachromatic leukodystrophy (MLD) is characterized by an inability to degrade sulfated glycolipids due to a deficiency of the lysosomal enzyme arylsulfatase A. The disorder affects central and peripheral myelin. There is widespread loss of myelinated oligodendroglia in the CNS and segmental demyelination of peripheral nerves (47). Clinically, it presents with motor regression and ataxia at around 2 years of age (47) and leads to a decerebrate state some years after the initial onset of symptoms. Epileptic seizures can occur late into the course of the illness. A late-onset form exists and presents in the third decade with psychiatric symptoms.
Earlier into the course of MLD, EEGs are often normal. If abnormalities are seen at presentation, they consist of mild slowing of the background activity (48). As the clinical severity of the disease progresses, so does the degree of slowing. Later into the course of the illness, as the patients develop epileptic seizures, focal spike waves appear. In the final stages of the disease course, hypersynchronous activity is seen (49). Both the clinical and EEG features of the disease can improve following bone marrow transplantation (50).
Globoid Leukodystrophy (Krabbe Disease)
Krabbe disease (KD) results from a deficiency of the lysosomal enzyme galactosylceramidase. Severe loss of myelin occurs and globoid cells are seen in the CNS white matter. Early infantile KD accounts for 90% of all cases and presents in the first 6 months of life (51) with irritability and rapidly progressive rigidity and tonic spasms. MRI of the brain shows symmetric signal abnormalities in the periventricular regions of the posterior cerebral hemispheres. Peripheral nerves are also affected, but no visceromegaly is observed. The remaining 10% of patients have late-infantile or juvenile KD, which presents with ataxia and later exhibits dystonia and visual failure (52).
EEGs are abnormal in a majority of the early infantile cases, but the opposite is true for late-infantile and juvenile-onset patients. Abnormalities consist of slowing without epileptiform activity in 46%, epileptiform activity without slowing in 15%, and both slowing and epileptiform features in 38% of patients (53). Likewise, BAEPs are abnormal in 88% of the early infantile patients and only 40% of the late-onset cases. The most frequent BAEP abnormality is absence of waves III and V (53).
PEROXISOMAL DISORDERS
Peroxisomal disorders are a group of heterogeneous conditions that share dysfunction of peroxisomal function. Peroxisomes are bound by a single membrane and contain a fine granular matrix. They are histologically identified by the presence of catalase. They are present in all human tissues except erythrocytes. They carry out over 40 metabolic functions, including beta-oxidation of very-long-chain fatty acids (VLCFA) and biosynthesis of plasmalogen and cholesterol. Some of those reactions can occur in other organelles, but many are exclusive to peroxisomes.
X-linked Adrenoleukodystrophy
X-linked adrenoleukodystrophy (X-ALD) is the most common of the peroxisomal disorders. It is characterized by accumulation of VLCFA in the brain and adrenal tissues (54). VLCFA are also detected in fibroblasts, blood cells, and plasma, allowing for identification of the condition (55). X-ALD affects the CNS or adrenal glands and can have diverse presentations. The cerebral form presents between 4 and 8 years of age with symptoms mimicking ADHD. Psychomotor regression and cortical blindness follow and seizures are observed late into the disease course. MRI shows lesions of the parieto-occipital white matter with contrast enhancement at the leading edge (56). The mean interval between the first symptom and an apparent vegetative state is 2.4 years (48). Adrenomyeloneuropathy is a variant that affects primarily the spinal cord and does not present until the third decade. Since X-ALD is an X-linked condition, it presents
earlier and with greater severity in males, but heterozygous women can be symptomatic.
earlier and with greater severity in males, but heterozygous women can be symptomatic.
During early stages of the disease, EEGs are normal in about half of patients and show posterior slowing in the remainder. The location of the slowing corresponds to the region of MRI abnormalities (48). As the disease progresses, the slowing becomes more widespread and paroxysmal discharges are observed (48). In one reported case, the ictal EEG during a tonic seizure revealed an electrodecrement of the background activity (48).
Zellweger Syndrome
Zellweger syndrome (ZWS), also known as cerebrohepatorenal syndrome, results from deficiencies of VLCFA beta-oxidation, phytanic acid oxidation, and plasmalogen synthesis. As a result, plasma levels of VLCFAs and phytanic acid are high, whereas erythrocyte concentrations of plasmalogen are reduced. ZWS presents in infancy with craniofacial dysmorphism (high forehead, large anterior fontanelle, separated cranial sutures, hypoplastic supraorbital ridges, and epicanthal folds) and hepatomegaly (57). Neurologic findings include hypotonia with absent reflexes, profound developmental delays, seizures, and impaired vision and hearing (57). MRI reveals both cortical (polymicrogyria and pachygyria) and white matter (delayed myelination) abnormalities (58).
Interictal EEGs in ZWS show bilateral independent multifocal spike waves, predominantly in the frontal motor cortex (59). Continuous negative vertex sharp and spike waves are commonly observed. They were found in 9 of 11 patients in one study (60) and seen in a single case in a separate report (61). This finding, observed during wakefulness and sleep, is pathognomonic for ZWS (61). BAEPs and VEPs can have absent or delayed responses (60).
Neonatal Adrenoleukodystrophy
Neonatal adrenoleukodystrophy (NALD) is a disorder of peroxysomal biosynthesis resulting from impaired ATPases that are necessary for importing proteins into peroxisomes. NALD presents at birth with facial dysmorphism (midface hypoplasia) (57). Hepatomegaly and adrenal cortical atrophy can also be observed. Neurologically, patients present with hypotonia, optic nerve atrophy, and seizures (62). Psychomotor regression occurs in early childhood. MRI reveals a severe deficiency of white matter in addition to heterotopias and polymicrogyria.
Interictal EEGs show high-voltage slow waves and bilateral independent multifocal spike waves. Later in the disease course, background suppression can be observed (59).
MITOCHONDRIAL DISORDERS
Mitochondrial diseases are a heterogeneous group of conditions that result from dysfunction of the mitochondrial respiratory chain. The mitochondria’s main function is generating ATP to power the cells they reside in. Because mitochondria are present in every cell type, with the sole exception of erythrocytes, mitochondrial dysfunction can have wide-ranging effects. The mitochondrial genome contains 37 genes, of which 13 encode structural proteins for the respiratory chain. The remaining 3000 or so genes are required to make a mitochondrion reside in the nuclear DNA. Inheritance patterns are therefore varied with both maternal and Mendelian patterns observed.
Leigh Syndrome
In 1951, Leigh described a case of subacute necrotizing encephalomyelopathy in a 7-month-old infant. The patient had symmetric lesions of the thalamus, midbrain, pons, medulla, and posterior columns of the spinal cord (63). Leigh syndrome results from defects in cytochrome oxidase or the pyruvate dehydrogenase complex. Inheritance patterns can be maternal, X-linked, or autosomal recessive. Patients presenting prior to 12 months are likely to exhibit psychomotor retardation, vomiting, weight loss, and weakness. In patients presenting after 18 months, symptoms include coma, ophthalmopareis, or nystagmus (64). Neuroimaging reveals abnormal signal in the basal ganglia and brain stem with sparing of the mamillary bodies. There is a strong predilection for the putamen; absence of putaminal involvement should call the diagnosis into question (65).
The interictal EEG can reveal focal or multifocal epileptiform activity, which becomes more frequent during sleep. During partial seizures, the ictal EEG reveals posterior or hemispheric background attenuation or ictal fast activity (66).
MELAS Syndrome (Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke)
MELAS is characterized by mitochondrial myopathy, encephalopathy, lactic acidosis, and recurrent stroke-like episodes. Many patients also exhibit seizures, migraines, cortical blindness, short stature, or ragged-red fibers on muscle biopsy (67). The mechanism of stroke in this condition remains controversial. Some implicate a mitochondrial angiopathy, resulting in small vessel occlusion, and others postulate that the impairment of oxidative metabolism results in lactic acidosis and brain injury (68). Imaging studies support the later hypothesis (68,69). MRI abnormalities are most pronounced in the parietal-occipital regions (69).
Likewise, interictal EEGs reveal diffuse sharp waves with an occipital predominance. The epileptiform abnormalities can be enhanced with photic stimulation (66). PLEDs can also be seen over the affected occipital lobe (70). In one report of 11 patients with a syndrome of overlapping MERRF-MELAS phenotype, EEG abnormalities correlated with the severity of the clinical phenotype. In milder cases, they included disorganization of the background activity and focal or generalized delta-theta waves (Fig. 15.2). More severely affected patients exhibited bursts of generalized spike or polyspike and slow-wave complexes (71). MELAS patients can develop epilepsia partialis continua. EEGs at those times reveal pseudoperiodic spike waves in the contralateral temporal-occipital regions (72).
Related posts:
 Dynamics of EEGs as Signals of Neuronal Populations: Models and Theoretical Considerations
Activation Methods
Metabolic Disorders and EEG
Anticipating Seizures Based on EEG
Intracranial Monitoring: Depth, Subdural, and Foramen Ovale Electrodes
EEG Event-Related Desynchronization (ERD) and Event-Related Synchronization (ERS)
Dynamics of EEGs as Signals of Neuronal Populations: Models and Theoretical Considerations
Activation Methods
Metabolic Disorders and EEG
Anticipating Seizures Based on EEG
Intracranial Monitoring: Depth, Subdural, and Foramen Ovale Electrodes
EEG Event-Related Desynchronization (ERD) and Event-Related Synchronization (ERS)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


