CHAPTER 176 Encephalocele
Encephalocele is herniation of cerebral tissue, meninges, and cerebrospinal fluid (CSF) outside the confines of the skull. The condition is also termed cephalocele to encompass any combination of these intracranial elements.1 Encephalocele has been recognized since antiquity; it was first depicted in ancient sculptures and later in medieval artwork to represent demonic creatures.2,3 Early written descriptions came from the Dutch physician Petrus Forestus (1590) and J.F.C. Corvinius (1749).2 Since then, numerous case reports and case series have been collected, the epidemiology and regional variations have been delineated, and the long-term outcomes of children treated in the latter portion of the 20th century have been documented. The past half century has witnessed advancements in the basic science of neural development, improvements in prenatal imaging and care, more global recognition of these developmental anomalies, and better access to quality care for children in developing countries, where the incidence of this condition is higher. This chapter presents our best understanding of the classification schemes, embryopathogenesis, epidemiology, management, outcomes, and prognosis for children with encephalocele.
Classification
Classification of encephaloceles is based on the anatomic location of the skull defect, as detailed in Table 176-1. The two main groups are anterior and posterior: anterior encephaloceles are divided into sincipital (from sinciput, or forehead) and basal, whereas posterior encephaloceles are divided into occipital, occipitocervical, and parietal. Detailed computed tomography (CT) and magnetic resonance imaging (MRI) in the modern era can demonstrate the precise location and anatomic contents of the herniation,4 thereby allowing further subclassification (see Table 176-1) than previously established through clinical experience.5
TABLE 176-1 Classification of Encephaloceles
| POSTERIOR ENCEPHALOCELES |
| ANTERIOR CRANIAL FOSSA ENCEPHALOCELES |
Embryology
The pathogenesis of encephalocele is poorly understood at a molecular level. Although encephalocele may represent defective closure of the neural tube in gross pathology, it does not appear to occur through defective neurulation. Primary neurulation, the process by which the future brain and the majority of the spinal cord form, occurs between the third and fourth gestational weeks through a process of embryonic folding and fusion of the midline (reviewed by Copp6) (Fig. 176-1). The initial closure point of the neural tube occurs at the region of the future occiput, whereas a second closure point takes place at the junction of the future forebrain and midbrain, with creation of a rostral (cranial) and caudal (spinal) neuropore. The rostral neuropore closes in a bidirectional fashion from the initial rhombencephalic, occipital closure (caudorostral), as well as in a rostrocaudal direction from a third closure site at the chiasmatic plate, with the extreme anterior end of the body axis corresponding to the future foramen caecum (see Fig. 176-1).7 As epithelial fusion occurs to create a neural tube, the dorsal midline tissues separate into an outer surface ectoderm (future epidermis) and inner neuroepithelium (future brain and spinal cord).8 Because encephaloceles are typically covered with normal skin, the initial closure event has already taken place, and thus the defect must arise after primary neurulation. Theories regarding encephalocele formation are focused on this stage of neural development, after the first month of gestation.
The most widely accepted theory is derived from that of Etienne Geoffroy Sain Hilaire (1827), the French naturalist whose work influenced Charles Darwin’s, which states that encephalocele is caused by an error in mesodermal differentiation.9 The paraxial mesoderm, from which the meninges and skull will form, migrates in between the ectodermal layers. In cases of encephalocele formation, a neuroschisis (fissure) develops after primary neurulation, which leads to scarring and subsequent adhesion between the cutaneous and the neuroectoderm and prevents interposition of the mesoderm. Pathologic specimens appear to follow these patterns in encephalocele formation, and a skull defect is present through which variably abnormal meninges and neural elements herniate.
Epidemiology and Further Theory Regarding Embryopathogenesis
The overall worldwide incidence of encephalocele is impossible to quantify because the majority of congenital encephaloceles lead to spontaneous abortion10 and collection of epidemiologic data in developing nations, where many of these lesions occur, is challenging.11 For unknown reasons, the prevalence of encephalocele in live births varies according to geographic location and race. In North America, Europe, and northern Asia, occipital encephaloceles predominate at a frequency varying between 0.8 and 4.0 per 10,000 live births, and sincipital encephaloceles are considerably more rare.10 By contrast, in Southeast Asia, sincipital encephaloceles are the predominant type and occur in 1 in 5000 live births, and occipital encephaloceles are much less common.12,13 Congenital basal encephaloceles represent just 10% of all encephaloceles in most series.14
Both remote and more recent epidemiologic data on the effect of seasonal variations and environmental exposure on the development of sincipital encephalocele have added credence to a toxicologic theory of this birth defect.13–15 A higher incidence of frontoethmoidal encephaloceles occurs in newborns conceived during the rainy season in Burma and Cambodia, when fungal molds are found in rice. A well-documented and ubiquitous fungal teratogen such as aflatoxin could combine with genetic factors to cause this condition.13,14 Most recently, a higher incidence of sincipital encephalocele was identified in a population of Assamese tea workers. An environmental factor such as a pesticide has been postulated to play a role.15 However, almost all congenitally acquired encephaloceles—and sincipital encephaloceles in particular—are sporadic, and familial association, though reported, is unusual.16 The genetic factors that would predispose an embryo to sincipital encephalocele are unknown.
Unlike sincipital encephaloceles, occipital encephaloceles are included in several rare but severe autosomal recessive and teratogenic syndromes. In syndromic cases, occipital encephaloceles are seen most commonly in Meckel-Gruber (or Meckel) syndrome (MGS), one of the cerebello-oculo-renal syndromes. Although MGS is a genetically heterogeneous disorder, proteins related to the syndrome mediate ciliary formation and epithelial morphogenesis at an early and critical developmental stage17 as a result of impairments in Sonic Hedgehog signaling.18 Impairments in neural tube patterning, governed by these processes, could in theory lead to the neuroschisis and subsequent failure of mesodermal interposition that occur after neural tube closure in St. Hilaire’s model of encephalocele formation. Beyond these scant details, the molecular pathogenesis of encephalocele formation remains unknown, but the details encoded in these developmental signaling pathways are certain to provide answers in the coming years.
Clinical Findings, Diagnosis, Management, and Outcome
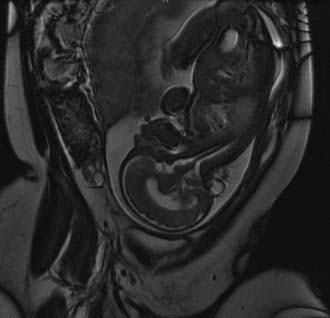
Despite their varied demographics and outcome, the principles of management of sincipital, cranial vault, and basal encephaloceles are similar and focus on removal of the sac, preservation of functional neural tissue, protection from leakage of CSF by closure of the meninges and nondysplastic skin, and correction of the cosmetic deformity. Prenatal diagnosis with amniocentesis (α-fetoprotein and acetylcholinesterase levels may vary from normal to abnormal, depending on epithelialization of the lesion), fetal ultrasound, and fetal MRI (Fig. 176-2) is commonplace today and allows accurate characterization of the location and type of encephalocele expected.
Postnatal MRI is the diagnostic study of choice to classify the encephalocele and predict the degree of functional tissue inside the sac and the extent of hydrocephalus, if any. Furthermore, concomitant magnetic resonance angiography/venography (MRA/MRV) may be useful in defining the lesion with respect to its surrounding vasculature. Volumetric (three-dimensional) CT can help in planning craniofacial reconstruction in infants with sincipital encephaloceles, although the need for such an extensive work-up has been called into question in recent series in locations where access to such studies is limited and surgical familiarity with the pathologies expected and encountered make them superfluous.11,19 Given the comparative rarity of sincipital encephaloceles in the Western hemisphere and the complex management issues that they present, a comprehensive work-up can certainly be warranted.
Posterior Encephaloceles
In the Western hemisphere, occipital encephaloceles are encountered most frequently, and with the advent of better prenatal imaging, they are becoming more frequently diagnosed during pregnancy.20 In the prenatal period, family counseling and a discussion of management and outcome must take place based on adequate imaging, knowledge of the historical and more recent literature concerning the long-term results after encephalocele repair10,20–22 (discussed later), and knowledge of the parents’ ethical and religious considerations.
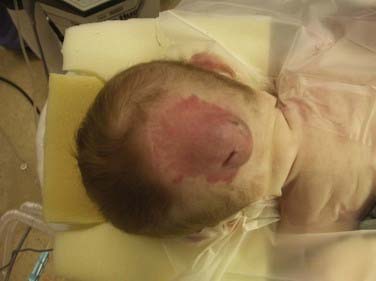
Occipital encephaloceles occur between the lambda and foramen magnum, typically in the midline, and are divided into supratorcular and infratorcular, depending on their proximity to the confluence of the sinuses. Occipitocervical encephaloceles incorporate a cranial and cervical spinal bony defect. Parietal encephaloceles exist at any point between and including the lambda and bregma. As mentioned earlier, well-detailed MRI and MRA sequences will show the involvement of neurovascular structures within or near the sac, associated hydrocephalus, and other related abnormalities in the craniocervical axis. Occipital encephaloceles vary in their appearance, size, and contents, from small, atretic lesions (Fig. 176-3) with little or no brain elements to large lesions (Figs. 176-4 to 176-6) that may have portions of functional cerebellum, cortex, and brainstem contained within them. Associated central nervous system findings in patients with occipital encephaloceles include hydrocephalus, kinking of the brainstem, and an absent, rudimentary, or inverted cerebellum with the brainstem herniated posteriorly and the cerebellar vermis ventrally. A small posterior fossa or posterior fossa cysts reminiscent of a Dandy-Walker variant, along with abnormal displacement of the torcular, transverse sinuses, and tentorium, may be seen. Cortical dysplasia and callosal agenesis are frequently present.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


