Video 42.1; see Chapter 6).
6. Juvenile absence epilepsy (see Chapter 6).
7. Juvenile myoclonic epilepsy (impulsive petit mal) of Janz (see Chapter 6).
8. Epilepsy with GTCS on awakening (see Chapter 6).
9. Generalized epilepsy with febrile seizures plus (GEFS+).
Autosomal-dominant disorder manifesting with febrile seizures in children <1 year of age, which persist beyond 5 to 6 years, when nonfebrile seizures, usually GTCS, also occur. Seizures may persist into adolescence or longer. Family history of febrile seizures is necessary to the diagnosis. It has been linked to a number of gene loci (SCN1B, SCN1A, and GABRG2). GEFS+ occurs with inherited missense mutations of SCN1A, while de novo truncating mutations result in severe myoclonic epilepsy of infancy (SMEI; Dravet’s syndrome).
B. Symptomatic or cryptogenic generalized epilepsy. These disorders, which are listed in order of age of appearance, include generalized epilepsy syndromes secondary to known or suspected disorders of the central nervous system (CNS; symptomatic) or to disorders, the causes of which are hidden or occult (cryptogenic).
1. Infantile spasms (West’s syndrome) (see Video 42.2).![]()
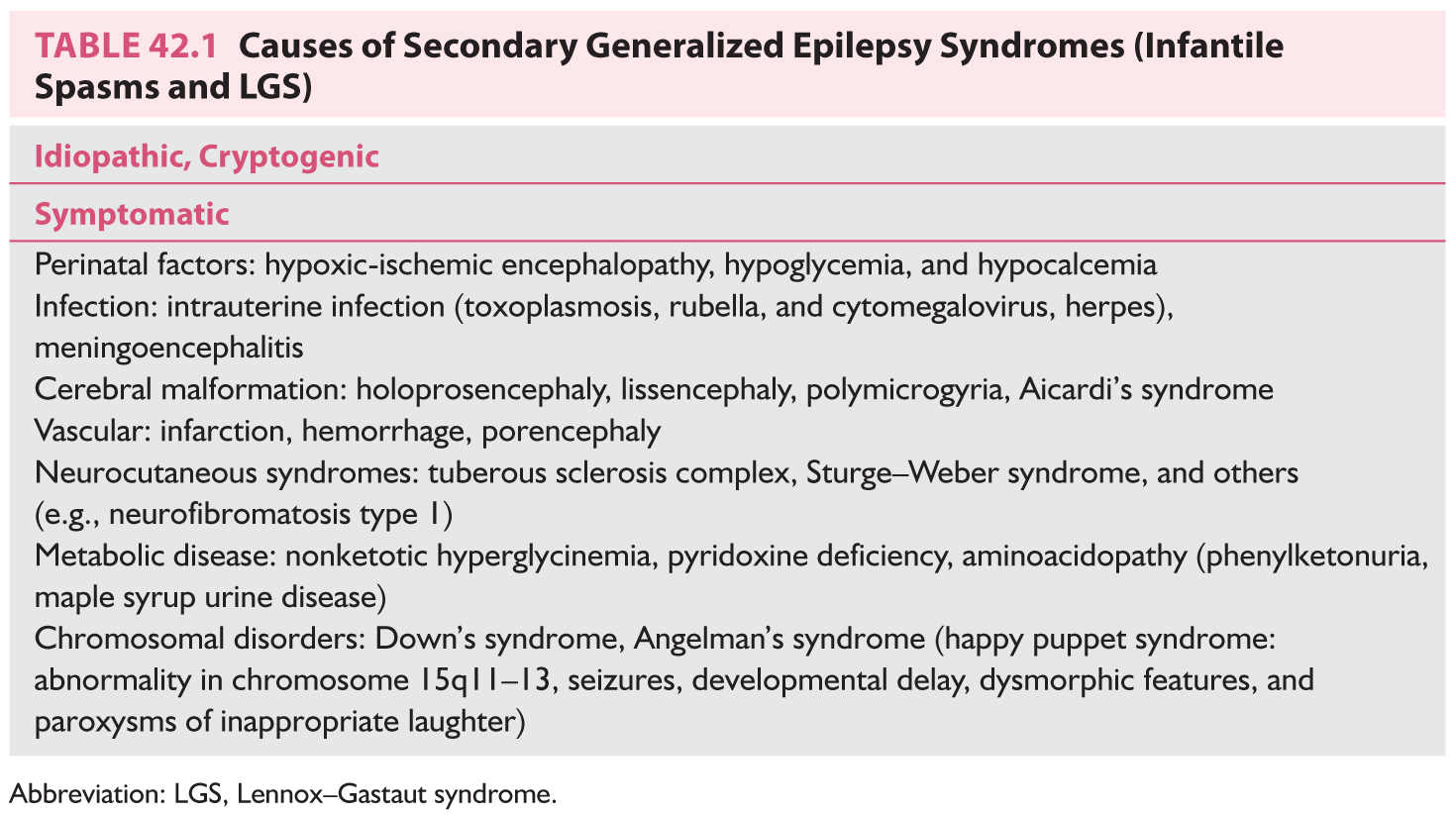
a. Etiology. With the availability of newer neuroimaging techniques, only 10% to 15% of cases are cryptogenic. In symptomatic cases, there is evidence of previous brain damage (mental retardation, neurologic and radiologic evidence, or a known etiologic factor) (Table 42.1).
b. Age at onset. Onset occurs in infancy (peak 4 to 8 months).
c. Clinical features compose the triad of infantile spasms, mental retardation, and hypsarrhythmia. Infantile spasms occur in clusters, frequently during drowsiness and on awakening, characterized by brief nodding of the head associated with extension or flexion of the trunk, and often of the extremities. They occur rapidly, suggestive of a startle reaction. They can be flexor (salaam attacks), extensor, or most commonly, mixed spasms. They are almost always associated with arrested development.
d. EEG shows hypsarrhythmia—chaotic, high-amplitude, disorganized background with multifocal independent spike-and-wave discharges and electrodecremental periods. Intravenous (IV) pyridoxine (vitamin B6) should be administered in a dose of 100 mg during the EEG to exclude pyridoxine-dependent infantile spasms.
e. Treatment.
(1) Underlying conditions are managed as identified. Adrenocorticotropic hormone (ACTH) and vigabatrin are the initial drugs of choice.
(2) ACTH. Opinions vary regarding dosage and duration of ACTH therapy, ranging from high-dose therapy (150 IU/m2/day) to low-dose therapy (20 to 40 IU/day). We recommend starting at 40 to 80 IU/day administered intramuscularly and continuing for 3 to 4 weeks, or for a shorter period if an early positive clinical response is observed. The dosage is then slowly decreased approximately 20% per week over 6 to 9 weeks. If seizures recur during withdrawal, the dosage should be increased to the previous effective level. ACTH therapy is initiated in the hospital under the guidance of a pediatric neurologist. Parents should be taught the injection technique with systematic rotation of the injection site.
(3) Side effects of ACTH therapy are irritability, hyperglycemia, hypertension, sodium and water retention, potassium depletion, weight gain, gastric ulcers, occult gastrointestinal bleeding, suppression of the immune system, infection, cardiomyopathy, congestive heart failure, and diabetic ketoacidosis.
(4) Laboratory tests before initiation of ACTH therapy include baseline EEG, serum electrolytes, blood urea nitrogen (BUN), serum creatinine, glucose, urinalysis, complete blood count (CBC), chest radiograph, and tuberculin skin test when appropriate.
(5) Laboratory tests performed weekly during ACTH therapy include serum electrolytes, blood glucose, stool guaiac, and monitoring of weight and blood pressure.
(6) Concomitant management. An antacid or a histamine H2 receptor antagonist (ranitidine) should be administered during ACTH therapy.
f. Alternative treatment.
(1) Prednisone may be substituted when ACTH cannot be administered because parents cannot or will not learn to give injections. It is administered orally at 2 to 3 mg/kg/day for 3 to 4 weeks and gradually withdrawn in a schedule similar to ACTH withdrawal.
(2) Other AEDs. Vigabatrin (100 to 150 mg/kg/day) has the best response rates in patients with tuberous sclerosis. However, irreversible constriction of peripheral vision may occur in 30% to 50% of patients. It is therefore administered for not more than 6 months, and discontinued in 3 weeks if no response is noted. Valproic acid (usually at high therapeutic levels of 75 to 125 mg/mL) is used cautiously because of a higher risk of liver dysfunction in children younger than 2 years of age. Topiramate, zonisamide, and clonazepam have also been reported to be effective. Nitrazepam and clobazam have also been tried.
(3) Excisional surgery of the region of cortical abnormality defined at EEG, magnetic resonance imaging (MRI), and positron emission tomography (PET) is being performed on children with infantile spasms intractable to medical therapy, but only in specialized centers.
g. Prognosis. West’s syndrome has a high morbidity, with a 90% incidence of mental retardation. From 25% to 50% of cases evolve into other type of epilepsies such as Lennox–Gastaut syndrome (LGS), with the infantile spasms transforming to other seizure types (GTCS, myoclonic, and tonic seizures) over subsequent years. Favorable prognostic indicators are as follows:
(1) Cryptogenic spasms have a better prognosis than symptomatic cases.
(2) Normal development before the onset of spasms and normal brain MRI
(3) Short duration of seizures before control
a. Etiology. A large number of patients have a history of infantile spasms. About 10% to 40% of cases are cryptogenic. In the 60% to 90% of symptomatic cases, a specific cause, usually perinatal insult, is found (Table 42.1).
b. Age at onset is 1 to 8 years of age, with peak between 3 and 5 years.
c. Clinical features are seizures of multiple types, typically tonic, atypical absence, and also atonic, and myoclonic seizures. GTCS and partial seizures may also be seen. Seizures are often frequent and intractable to medical treatment. Most patients have cognitive dysfunction.
d. EEG shows slow background activity, generalized, bisynchronous, 2 to 2.5 Hz spike and sharp–slow-wave discharges activated by sleep, generalized paroxysmal fast spike activity (10 Hz), and other multifocal epileptiform abnormalities.
e. Treatment.
(1) AEDs. Broad-spectrum AEDs such as valproic acid are required for treatment against all the different types of seizures associated with LGS. However, these seizures are often intractable, and valproic acid may have to be used in combination with other broad-spectrum AEDs. Ethosuximide, lamotrigine, and topiramate have successfully demonstrated efficacy as adjunctive therapy in LGS. Rufinamide, zonisamide, and levetiracetam have also been reported to be effective. Felbamate, though effective, is infrequently used because of severe side effects such as aplastic anemia and acute liver failure. Phenytoin and phenobarbital may be helpful in controlling the associated GTCS. Benzodiazepines (clonazepam, nitrazepam, and clobazam) can be used, but may be associated with side effects of decreased alertness and drowsiness, which are associated with increased seizure frequency. IV diazepam or lorazepam may induce tonic seizures, and carbamazepine can exacerbate absence seizures.
(2) Ketogenic diet may be effective for patients with otherwise intractable seizures. Benefits include fewer seizures, less drowsiness, and fewer concomitant AEDs.
(3) ACTH has been found to be effective in the treatment of some patients.
(4) Psychological support for the child and family. A prescription for protective helmets to prevent head injuries in patients with drop attacks is helpful.
(5) Surgical procedures such as corpus callosotomy, hemispherectomy, and rarely resection of a localized lesion have been tried with variable results. Vagal nerve stimulation is also effective with at least 50% reduction in seizure frequency in follow-up periods as long as 5 years.
3. Myoclonic astatic epilepsy (MAE)—Doose’s syndrome.
a. Age at onset is from 6 months to 6 years with a peak at 3 years, with male preponderance.
b. Clinical features. GTCS are seen at onset, progressing to the characteristic myoclonic or myoclonic astatic seizures characterized by a myoclonic jerk followed by loss of muscle tone, which may result in a sudden fall. Absence seizures are seen as well, often associated with a clonic component. Seizures occur frequently on awakening. Tonic seizures are less common in contrast to LGS.
c. EEG may be initially normal except for brief bursts of parietal 4 to 7 Hz theta activity. Brief bursts of generalized spike and polyspike wave discharges are seen later.
d. Treatment. Valproic acid is the drug of choice. Ethosuximide and ketogenic diet are also effective.
e. Prognosis ranges from remission of seizures with normal development to intractable epilepsy with severe cognitive impairment.
4. Symptomatic seizures. Myoclonic seizures are difficult to differentiate from nonepileptic myoclonus. However, characteristic epileptiform discharges associated with myoclonic jerks in myoclonic epilepsy help differentiate the two.
a. Early myoclonic encephalopathy is characterized by the onset of medically intractable myoclonic seizures and partial seizures in early infancy before 3 months of age, burst suppression on EEG, and very poor prognosis including profound neurologic impairment or death in the first year of life. Multiple causes include inborn errors of metabolism such as nonketotic hyperglycinemia, methymalonic acidemia, and proprionic acidemia.
b. Early infantile epileptic encephalopathy (Ohtahara’s syndrome) is characterized by an early onset of tonic spasms within the first few months of life, which are medically intractable. Myoclonic seizures are rare. The burst-suppression pattern on EEG is present during waking and sleep states. MRI demonstrates severe developmental anomalies such as hemimegalencephaly, porencephaly, and Aicardi’s syndrome. The prognosis is very poor. Mutations in the gene encoding syntaxin binding protein 1 (STXBP1) have been reported.
c. Severe Myoclonic Epilepsy in Infancy (Dravet’s syndrome) represents 3% to 5% of all epilepsies starting in the first year of life. The disorder begins in the first year of life as febrile seizures, followed by myoclonic seizures, atypical absences, and convulsive seizures between 1 and 4 years of age. The seizures are often triggered by fever, hot baths, or hot weather. The child is initially normal, but cognition becomes progressively impaired. EEG shows generalized spike and polyspike–slow wave activity, focal or multifocal spikes. Photosensitivity is seen in 40%. Sodium-channel blockers such as carbamazepine, oxcarbazepine, phenytoin, and lamotrigine may induce worsening of seizures. The seizures are medically intractable, but may respond to valproic acid, topiramate, and clobazam. Stiripentol has also proved to be effective as is ketogenic diet. A link between SMEI and GEFS+ has been identified in several families. SCN1A gene mutations (both truncating and missense) on chromosome 2p24 in coding for the neuronal voltage-gated sodium channel a1 subunit have been found in 80% of patents with SMEI, and 95% of these are de novo. Prognosis is poor with persistent severe developmental delay and intractable seizures.
d. Symptomatic myoclonic epilepsy is associated with specific progressive neurologic diseases such as Lafora’s disease, Baltic myoclonus (Unverricht–Lundborg disease), neuronal ceroid lipofuscinosis (Batten’s disease), sialidosis, mitochondrial encephalomyopathy, and Ramsay–Hunt syndrome.
EPILEPSIES AND SYNDROMES UNDETERMINED WHETHER FOCAL OR GENERALIZED
A. Neonatal seizures. Seizures occur most frequently in the neonatal period than at any other time in life, with an incidence of 1.5 to 5.5 per 1,000 live births.
1. Clinical features. Neonatal seizures (occurring between birth and 2 months) are more fragmentary than are seizures among older children. GTCS do not occur in neonates. Common causes are outlined in Table 42.2. Neonatal seizures are classified as follows:

a. Seizures associated with electrographic signatures include focal and multifocal clonic seizures, focal tonic seizures, generalized myoclonic seizures, and, rarely, apnea. These seizures are usually associated with focal structural lesions (infarction or hemorrhage), infection, or metabolic abnormalities (hypoglycemia or hypocalcemia).
b. Seizures not associated with electrographic signatures include generalized tonic seizures, focal and multifocal myoclonic seizures, and subtle seizures (oral–buccal–lingual movements, bicycling movements, and some rhythmic ocular movements such as horizontal eye deviation). These seizures are usually observed among lethargic, comatose neonates with poor prognoses, such as those with severe hypoxic-ischemic encephalopathy.
2. Evaluation. Neonatal seizures should be managed in a neonatal intensive care unit by experienced personnel, including a pediatric neurologist and a neonatologist.
a. History and examination. History of maternal illness, infection, or drug and alcohol abuse during pregnancy should be obtained. Family history of neonatal seizures is suggestive of BFNS. Evaluation of the skin, anterior fontanel, and neurologic and ophthalmologic examinations should be performed. Presence of skin rash and chorioretinitis may suggest toxoplasmosis.
b. Laboratory data include CBC, serum glucose, electrolytes, BUN, serum creatinine, liver function tests, magnesium, calcium, phosphate, ammonia, lactate, pyruvate, biotinidase, lumbar puncture (LP) to rule out CNS infection and subarachnoid hemorrhage (SAH), titers for toxoplasmosis, rubella, cytomegalovirus, herpes (TORCH), and HIV. Additional studies such as plasma amino acids and very-long-chain fatty acids, urinalysis for amino acids and organic acids, cerebrospinal fluid (CSF) lactate, and neurotransmitters may be indicated if metabolic disorders are suspected. Ultrasonography of the head at bedside to rule out intracranial hemorrhage and a non-contrast-enhanced computed tomography (CT) or MRI of the head can be performed when the neonate’s condition is stable. EEG is useful for the diagnosis of subclinical seizures and assessment of prognosis. EEGs with low-voltage, burst-suppression, or isoelectric patterns suggest poor prognosis.
3. Treatment.
a. Management of underlying cause such as CNS infection or specific metabolic abnormality (hypoglycemia, hypocalcemia, or hypomagnesemia)
b. Phenobarbital, the initial drug of choice, is administered IV as a loading dose of 20 mg/kg followed by additional 5 to 10 mg/kg boluses as required to achieve serum levels between 20 and 40 mg/mL and to control clinical seizures. Maintenance dose of 3 to 4 mg/kg/day is usually sufficient because phenobarbital has a relatively long half-life in neonates. Cardiorespiratory monitoring is important because IV administration can be associated with respiratory depression and hypotension.
c. Fosphenytoin is added if a phenobarbital level of 40 mg/mL is not sufficient to control seizures. An IV loading dose of 20 mg/kg phenytoin equivalent (PE) results in serum levels ranging from 15 to 20 mg/mL, followed by a maintenance dose of 3 to 4 mg/kg/day. Phenytoin is no longer used because it has to be infused slowly at 1 mg/kg/minute with cardiac monitoring, as it can cause cardiac arrhythmias and hypotension. It is alkalotic and may lead to local venous thrombosis or tissue irritation. Use of fosphenytoin is preferred because it reduces these risks.
d. Benzodiazepines are third-line treatments. Midazolam is usually the benzodiazepine of choice. It is administered IV at a loading dose of 0.15 mg/kg followed by 0.04 to 0.4 mg/kg/hour infusion. Lorazepam 0.05 to 0.1 mg/kg administered IV enters the brain rapidly, being effective in <5 minutes. Being less lipophilic, it does not redistribute from the brain as rapidly as does diazepam, so the duration of action is longer, being 6 to 24 hours. Lorazepam is less likely to produce respiratory depression or hypotension than diazepam.
e. Newer AEDS. Levetiracetam and topiramate are also used for refractory neonatal seizures. IV levetiracetam has been advocated by some, as a second line of treatment instead of fosphenytoin, if phenobarbital is ineffective.
f. Pyridoxine (100 mg IV) administered during EEG monitoring stops seizures and normalizes the EEG within minutes in the rare patient with pyridoxine-dependent seizures.
B. Acquired epileptic aphasia (Landau–Kleffner syndrome) is characterized by acquired aphasia, with onset between 3 and 7 years, including verbal auditory agnosia, rapid reduction of spontaneous speech, and behavioral and psychomotor disturbances, during the first decade of life. Seizures (generalized and focal) and EEG abnormalities, including multifocal spikes and spike–wave discharges commonly in the temporal or parieto-occipital regions, are activated by sleep. The ultimate outcome is still unclear.
A. Situation-related seizures.
1. Febrile seizures.
a. Incidence. Febrile seizures occur in 2% to 5% of young children.
b. Age at onset ranges from 3 months to 5 years (peak 18 months to 2 years). The disorder is familial.
c. Clinical features manifest within the first few hours of acute infection, usually associated with the rising phase of the temperature curve. They are typically associated with viral upper respiratory tract, gastrointestinal, and middle ear infections. Bacterial infection is rarely associated with febrile seizures. Intracranial infection and other defined causes such as dehydration and electrolyte imbalance should be excluded.
(1) Simple febrile seizures present as single, brief (<15 minutes), GTCS. They represent 80% to 90% of all febrile seizures, and are usually seen in neurologically normal children.
(2) Complex febrile seizures are prolonged (>15 minutes), have focal features (focal onset or postictal Todd’s paralysis), and more than one seizure occurs within 24 hours.
d. Evaluation. LP is indicated unless the possibility of meningitis can be confidently eliminated clinically. It should be performed for all children younger than 18 months with the first febrile seizure. If in doubt, err on the side of performing LP. It should be strongly considered in the evaluation of infants and children who have received antibiotic treatment because such treatment can mask evidence of meningitis. Neuroimaging and EEG are indicated if there are focal deficits or CNS infection is suspected.
e. Acute management of seizures.
(1) Prolonged febrile seizures can be treated with IV lorazepam, diazepam, or phenobarbital.
(2) Rectal diazepam gel (Diastat 0.2 to 0.5 mg/kg) can be administered at the onset of a seizure in children with history of prolonged seizures.
(3) Any underlying infection or fever should be controlled.
f. Long-term management of seizures. No treatment is necessary if the patient has isolated simple febrile seizures without major risk factors for recurrence.
(1) Daily phenobarbital treatment reduces the risk of recurrent febrile seizures and may be used for patients with complex febrile seizures that carry increased risk of later epilepsy. Because 90% of febrile seizures recur within 2 years, treatment should be continued for at least 2 years or for 1 year after the last seizure, whichever is longer. Valproic acid is the second choice because of an increased incidence of side effects, including liver toxicity, in this age group. Carbamazepine and phenytoin do not prevent recurrent febrile seizures.
(2) Some pediatric neurologists use oral diazepam, administered only when fever is present, and have found it effective in reducing the risk of recurrent febrile seizures. Side effects include lethargy, irritability, and ataxia.
g. Prognosis. Approximately 33% of children with febrile seizures have at least one recurrence, and 9% have three or more seizures. Remission occurs by 6 years of age in approximately 90% of children.
(1) Risk factors for recurrence include young age (<1 year) at initial seizure, seizures occurring with low-grade fever, and a family history of febrile seizures.
(2) Risk factors for the development of epilepsy include complex febrile seizures, underlying developmental or neurologic abnormalities, and family history of nonfebrile seizures. Three percent to six percent of children with febrile seizures will develop epilepsy. Prolonged febrile seizures may lead to temporal lobe epilepsy with hippocampal sclerosis. Patients with two risk factors have approximately 13% chance of developing epilepsy, whereas the risk is only 0.9% if risk factors are absent.
2. Seizures related to identifiable situations such as use of drugs (stimulants or neuroleptics) or alcohol, and sleep deprivation.
B. Isolated, apparently unprovoked epileptic events. Treatment is not indicated unless there are significant risk factors for recurrence.
C. Epilepsy characterized by specific modes of seizure precipitation includes seizures occurring in response to discrete or specific stimuli (reflex epilepsy), such as reading epilepsy, hot water epilepsy, and arithmetic epilepsy.
D. Chronic progressive epilepsia partialis continua of childhood (Kojewnikoff’s syndrome) is thought to be a result of chronic encephalitis (Rasmussen’s encephalitis). The cause is unknown. It is characterized by partial motor seizures, often associated with myoclonus, which are resistant to treatment. This condition results in progressive hemiplegia with unilateral brain atrophy and mental retardation.
EVALUATION
Details regarding histories, physical examinations, and studies such as EEG and neuroimaging are discussed in Chapter 6. Important aspects of the evaluation with respect to children follow. Determine whether the paroxysmal events in question are in fact epileptic. They should be differentiated from nonepileptic paroxysmal events in children.
SPECIFIC PREDISPOSING FACTORS FOR CHILDHOOD SEIZURES
A. Birth history.
1. Prenatal. Prematurity, complications (e.g., toxemia or premature labor), medications, smoking, and alcohol and drug abuse
2. Perinatal. Low birth weight, low Apgar scores, and complications of labor and delivery
3. Postnatal. Intensive neonatal respiratory care, complications such as meningitis and intraventricular hemorrhage
B. Developmental history. Learning disabilities, attention deficit, and developmental regression
PHYSICAL EXAMINATION
A. Head circumference. Microcephaly and macrocephaly are associated with various neurologic disorders.
B. Height and weight abnormalities
C. Dysmorphic features associated with chromosomal anomalies, storage diseases, or brain malformations
D. Skin. Café au lait spots are seen in neurofibromatosis, hypopigmented macules and adenoma sebaceum in tuberous sclerosis, and facial hemangiomas in Sturge–Weber syndrome.
E. Hair. Broken hair and alopecia suggest metabolic disorders (biotinidase deficiency, Menkes’ syndrome, and argininosuccinic aciduria).
F. Mental status and developmental milestones. Loss of previously attained milestones may be indicative of a neurodegenerative disease (e.g., Rett’s syndrome), whereas delays in achieving developmental milestones reflect static encephalopathies (e.g., cerebral palsy). Presence of anxiety, depression, and stressors such as family conflict may lead to the diagnosis of psychogenic seizures.
G. Systemic exam. Organomegaly may suggest a storage disease or an inborn error of metabolism.
LABORATORY TESTING
In addition to EEG and MRI of the head, other important studies in children include the following:
A. Chemical and metabolic screening. Electrolytes, glucose, calcium, magnesium, hepatic and renal function tests, and toxic screening for possible drug ingestion. Specific metabolic or neurodegenerative disorders may be diagnosed with tests such as thyroid function tests, urinalysis for amino acids, organic acids, homocysteine (cobablamin G deficiency and methylenetetrahydrofolate reductase deficiency), lysosomal enzymes (mucopolysaccharidosis and Batten’s disease), and very-long-chain fatty acids (peroxisomal disorders such as adrenoleukodystrophy). Elevation of serum prolactin levels is seen with GTCS, may be normal with focal seizures, and can help differentiate seizures from nonepileptiform paroxysmal disorders. However, it is not routinely performed because it has to be measured within 30 minutes after an episode and compared with baseline values.
B. LP is indicated if there are signs of acute CNS infection or inflammation (e.g., fever or stiff neck). It is indicated for all children younger than 18 months with a history of fever and seizures, because clinical signs of CNS infection may be absent. It should be performed on all febrile patients with new-onset seizures. A low CSF glucose with normal CSF cell count and protein and normal serum glucose suggests glucose transporter 1 deficiency (GLUT 1 deficiency), which can be confirmed with genetic testing of SLC2A1.
C. Chromosomal analysis is indicated if dysmorphic features suggest chromosomal abnormalities.
D. Genetic testing. Mutations in SCN1A have been associated with SMEI and GEFS+, and mutations in SCN1B, with GEFS+. Mutations in KCNQ2 gene have been associated with BFNC. Ohtahara’s syndrome has been associated with STXBP1 and infantile spasms with ARX. Children with epileptic encephalopathy (early-onset intractable seizures, developmental delay, cognitive impairment) should have chromosome microarray or targeted sequencing panels of epileptic encephalopathy genes including SCN1A, SCN1B, SCN8A, KCNQ2, STXBP1, and PCDH19.
E. Simultaneous prolonged video EEG monitoring in an epilepsy unit can help determine the exact nature of paroxysmal events if they cannot be defined with routine EEG.
TREATMENT
SINGLE SEIZURE
Approximately 9% of the population has a seizure sometime during their lives, and approximately 3% have more than one seizure. The first unprovoked seizure is defined as a single seizure or flurry of seizures within 24 hours in a child older than 1 month without prior history of unprovoked seizures. The risk of recurrence is 27% to 52%. It is highest in the first 6 months after the initial event. It is low if the patient has normal findings at neurologic examination, a single GTCS with a negative family history, normal findings at neuroimaging, and a normal or mildly slow EEG. It is higher if there is underlying structural or metabolic etiology.
A. Indications for treatment.
1. Clear-cut epileptiform abnormalities at EEG
2. Lesions on neuroimaging studies
3. Abnormal findings at neurologic examination that suggest previous brain damage
4. First seizure during sleep
5. Active CNS infection (encephalitis, meningitis, or abscess)
6. Certain types of seizures, including infantile spasms, LGS, and focal seizures
7. Unprovoked or asymptomatic single seizure with history suggesting that one may have occurred earlier
B. Treatment is not indicated when seizures are provoked by a correctable metabolic disturbance (glucose or electrolyte abnormalities), sleep deprivation, exposure to drugs or alcohol, febrile illness, or physical or emotional stress. In such cases, the underlying disturbance should be corrected.
GENERAL PRINCIPLES OF TREATMENT
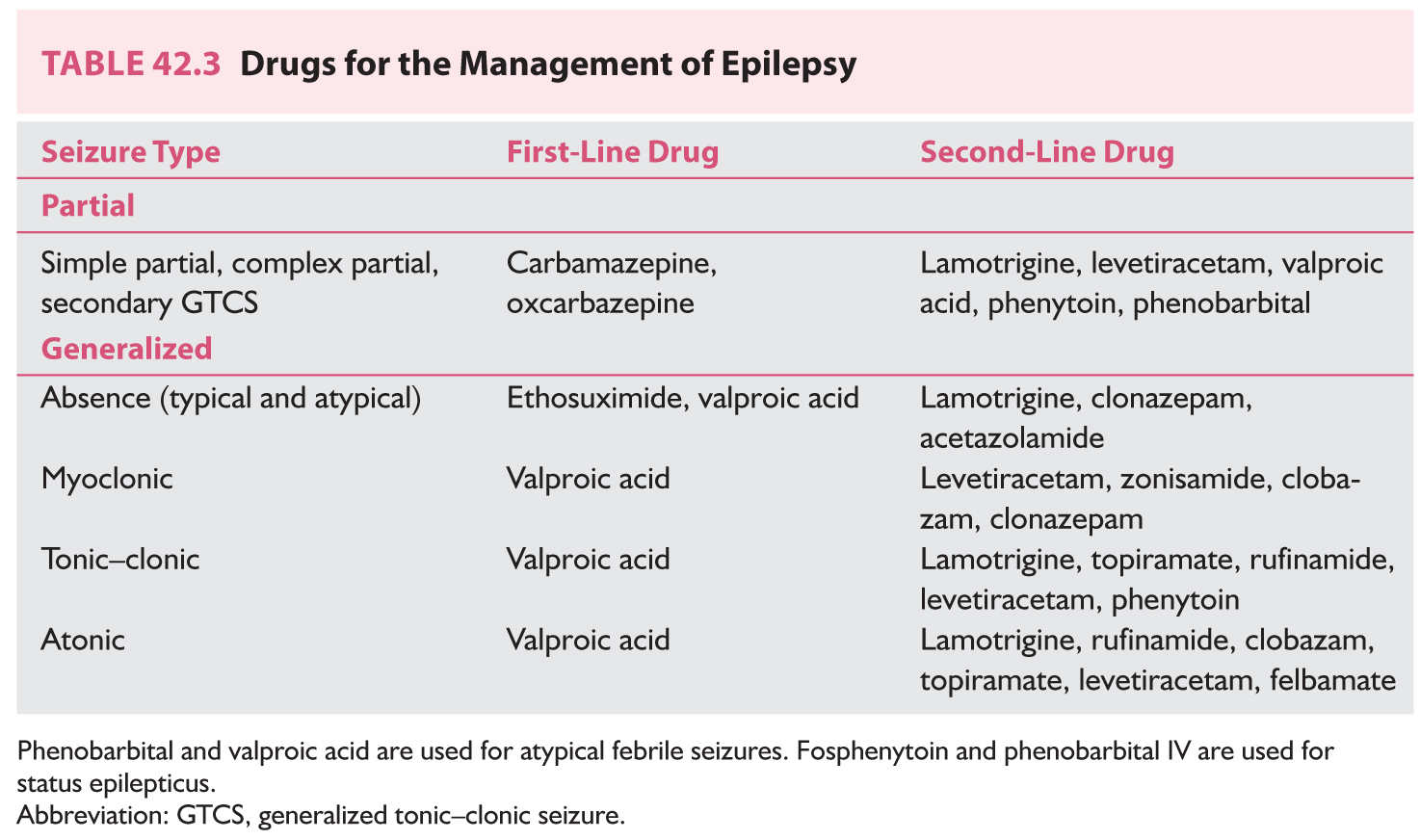
A. Choice of appropriate drug should be based on the clinical description of the seizures (Table 42.3). This choice may be influenced by other factors, such as the patient’s age, associated medical illnesses, and economic circumstances.
B. Monotherapy. Approximately 50% of children should respond to monotherapy with the first drug and another 15% to monotherapy with the second drug. If the first drug is ineffective, start a second AED with a different mechanism of action and low potential for adverse effects and drug interactions. After therapeutic levels are achieved, gradually withdraw the first drug.
C. Polypharmacy is indicated only if monotherapy with at least two first-line AEDs fails, and should be initiated only after consultation with a pediatric neurologist. Associated problems include drug interactions, difficulties in acquiring therapeutic levels of either drug despite use of adequate doses, increased risks of toxicity, increased cost, and reduced compliance.
D. Simplify medication schedule. Decreasing the number of doses improves compliance.
E. Avoid sedative anticonvulsants such as benzodiazepines, especially for patients with secondary generalized epilepsy syndrome, because increased sedation can result in increased seizures.
F. Maintain a seizure diary. Record seizure frequency, medication dosages and levels, and occurrence of side effects, if any.
G. AED level should be checked, preferably before the morning dose to obtain the lowest (trough) level and at a consistent time to avoid misinterpretation of fluctuations. CBC and aspartate aminotransferase levels are checked every 1 to 2 months initially and then every 6 months after a steady dosage has been established. AED blood level should be checked.
1. After starting a medication, to aid in initial titration of dose to achieve a therapeutic level
2. After making a major change in drug dosage
3. If seizures persist despite “correct therapy”
4. If symptoms of toxicity develop
5. If noncompliance is suspected
H. Repeat EEG during therapy if there is a change in the character of the seizures or if the child has been seizure-free for a considerable period to help decide whether medications can be withdrawn.
DRUG THERAPY
The following information serves as a broad guideline for AEDs commonly used to treat children. Indications are listed in Table 42.3. Details regarding their metabolism, side effects, and interactions are discussed in detail in Chapter 43.
A. Phenytoin (Dilantin; Parke-Davis).
1. Administration (5 to 8 mg/kg/day). Children have shorter elimination half-lives than adults and should be given two divided doses. Phenytoin has nonlinear elimination kinetics. Therefore, small increases in dose after therapeutic levels of 10 to 20 mg/mL have been achieved resulting in large increases in plasma levels and toxicity.
2. Formulation. Capsules: Dilantin, 30 mg, 100 mg; Infatabs: 50 mg; Phenytoin 100 mg, 200 mg, 300 mg (extended release); suspension: 125 mg/5 mL, 30 mg/5 mL. Dilantin suspension is not recommended for routine use because it is unreliable. Parenteral: injectable sodium phenytoin (Dilantin and generic 50 mg/mL), fosphenytoin [Cerebyx (Pfizer) 50 mg PE/mL].
3. Side effects. Dose-related side effects are nystagmus, ataxia, and drowsiness. Gingival hypertrophy (20% to 50% of patients) requiring more frequent dental cleaning, hirsutism, coarsening of features, blood dyscrasias, Stevens–Johnson syndrome, lymphadenopathy, systemic lupus erythematosus, and megaloblastic anemia can also occur. Long-term administration has been associated with vitamin D, vitamin K, and folic acid deficiency. Fetal hydantoin syndrome is characterized by craniofacial anomalies, hypoplasia of distal phalanges, intrauterine growth retardation, and mental deficiency. The term has been replaced by “fetal anticonvulsant syndrome,” because the malformations are also seen in children of mothers who have been exposed to a variety of other AEDs such as valproic acid, carbamazepine, and phenobarbital as well.
B. Carbamazepine (Tegretol, Tegretol XR; Novartis. Carbatrol, Shire–Richwood).
1. Administration. Start at 5 mg/kg/day and increase by 5 mg/kg/day every 5 to 7 days to a maximum of 10 to 30 mg/kg/day in bid (for sustained-release preparations) and tid doses to achieve therapeutic blood levels of 4 to 12 mg/mL. Carbamazepine can exacerbate absence, atypical absence, and myoclonic seizures.
2. Formulation. Tablets: 200 mg; chewable tablets: 100 mg; elixir: 100 mg/5 mL; sustained-release preparations: Tegretol XR: 100 mg, 200 mg, 400 mg; Carbatrol extended-release capsules: 200 mg, 300 mg. If oral administration is contraindicated, Tegretol suspension (100 mg/5 mL) can be given rectally, diluted 1:1 with water in an enema at a dose of 10 to 30 mg/kg to attain therapeutic levels.
3. Side effects. Dose-related side effects are sedation, blurred vision, and leukopenia. Agranulocytosis, aplastic anemia, and syndrome of inappropriate antidiuretic hormone secretion also may occur. There is a 0.5% to 1% risk of spina bifida and other anomalies associated with the fetal anticonvulsant syndrome, with first-trimester exposure to carbamazepine.
C. Oxcarbazepine (Trileptal; Novartis).
1. Administration. 10 mg/kg/day, to be increased by the same amount weekly to 20 to 40 mg/kg/day, in two divided doses to achieve therapeutic blood level of 10 to 35 mg/L. It has less potential for drug interactions because of lack of auto-induction.
2. Formulation. Tablets: 150 mg, 300 mg, 600 mg; suspension: 300 mg/5 mL.
3. Side effects. Somnolence, dizziness, and headaches. Cross-allergy between carbamazepine and oxcarbazepine occurs in 35% of cases. Hyponatremia is more frequent than with carbamazepine, seen in 2.5% of patients.
D. Phenobarbital (various generic formulations).
1. Administration. 3 to 8 mg/kg/day in a single daily dose or two divided doses to achieve therapeutic blood levels of 15 to 40 mg/mL. The serum half-life increases with age. It is 20 to 65 hours for patients younger than 10 years and 64 to 140 hours for those older than 15 years. Therefore, children need higher maintenance dosages of 4 to 8 mg/kg/day; adults need only 1 to 2 mg/kg/day. It can exacerbate absence, atypical absence, and myoclonic seizures, if used for GTCS in patients with generalized epilepsy.
2. Formulation. Tablets: 15 mg, 16.2 mg, 30 mg, 32.4 mg, 60 mg, 64.8 mg, 97.2 mg, 100 mg; elixir: 20 mg/5 mL. Parenteral: injectable sodium phenobarbital (60 mg/mL, 130 mg/mL).
3. Side effects. Hyperactivity, sedation, learning disabilities, personality changes, and Stevens–Johnson syndrome. Long-term administration has been associated with vitamin D, vitamin K, and folic acid deficiency.
E. Valproic acid (Depakene, Depakote, Depacon; Abbott).
1. Administration. Start at 10 to 15 mg/kg/day or lower, and gradually increase to a maximum of 60 mg/kg/day in three divided doses to achieve therapeutic blood levels of 40 to 100 mg/mL.
2. Formulation. Depakote (divalproex sodium): enteric-coated tablets, 125 mg, 250 mg, 500 mg; sprinkles, 125 mg; extended-release tablets, 250 mg, 500 mg. Depakene (valproic acid): capsules, 250 mg; syrup, 250 mg/5 mL. Parenteral IV preparation: Depacon (100 mg/mL) starting at 10 to 15 mg/kg/day increased 5 to 10 mg/kg/day/week to a maximum of 60 mg/kg/day infused over 60 minutes at a rate not to exceed 20 mg/minute. If oral administration is contraindicated (e.g., paralytic ileus), Depakene elixir (250 mg/5 mL) can be given rectally, diluted 1:1 with water in an enema at a dose of 20 mg/kg to attain therapeutic levels of 40 to 100 mg/mL.
3. Side effects. Dose-related side effects include nausea, vomiting, and gastric irritation, minimized with the use of the sprinkle or enteric-coated preparation or administration after meals. Other side effects include weight gain, alopecia, tremor, and thrombocytopenia. Idiosyncratic toxicity includes pancreatitis (0.5%) and liver failure. Liver failure is more common among children younger than 2 years, and with polypharmacy. It can be a fulminant progressive failure or a subacute gradually progressive failure. Valproic acid is therefore contraindicated in the treatment of children with preexisting hepatic damage, organic aciduria, or carnitine deficiency. Carnitine (Carnitor, 10% solution or 330 mg tablet) should be administered 50 mg/kg/day in two or three divided doses in conjunction with valproic acid to children undergoing long-term, high-dose therapy with poor nutrition (e.g., cerebral palsy). Baseline liver function and serum ammonia should be checked before starting valproic acid and at least monthly for the first 4 to 6 months while this medication is being given. It may be associated with increased risk of polycystic ovarian syndrome.
F. Lamotrigine (Lamictal; GlaxoSmithKline).
1. Administration. Coadministration of valproic acid increases the elimination half-life of lamotrigine to 60 hours or more. Therefore, the dose needs to be adjusted. Children not taking valproic acid: initial dose of 0.6 mg/kg/day for 2 weeks, increased to 1 mg/kg/day for 2 weeks and thereafter, slowly titrated to a maximum of 5 to 15 mg/kg/day twice a day. Children taking valproic acid: initial dose of 0.15 mg/kg/day for 2 weeks, increased to 0.3 mg/kg/day for 2 weeks and slowly titrated to a maximum of 1 to 5 mg/kg/day twice a day. Therapeutic blood levels are 5 to 15 mg/mL.
2. Formulation. Chewable dispersible tablet: 2 mg, 5 mg, 25 mg; tablets: 25 mg, 100 mg, 150 mg, 200 mg.
3. Side effects. Common adverse effects among children include somnolence, rash, vomiting, laryngitis, ataxia, and headache. Risk of skin rash in children is 1 in 100 to 200 as compared with 3 in 1,000 in adults and is seen within the first 6 weeks. The incidence of rash increases with higher initial doses and faster rates of dose escalation, especially among children receiving valproic acid.
G. Topiramate (Topamax; Ortho-McNeil).
1. Administration. Initiated at 0.5 to 1 mg/kg/day increased slowly to 4 to 9 mg/kg/day, given in two divided doses. Faster titration schedules have resulted in increased CNS side effects. Therapeutic blood levels are 3.4 to 16.6 mg/mL.
2. Formulation. Tablets: 25 mg, 50 mg, 100 mg, 200 mg; sprinkle capsule: 15 mg, 25 mg.
3. Side effects. Somnolence, dizziness, ataxia, psychomotor slowing, speech disorder, paresthesia, kidney stones (1.5%), and weight loss. Decreased sweating usually with exposure to hot weather occurs more frequently in children. Acute myopia with secondary angle closure glaucoma has been reported, usually within the first month. New data suggest a higher risk of cleft palates in babies born to women taking the drug.
H. Levetiracetam (Keppra; UCB Pharma).
1. Administration. Initiated at 10 to 20 mg/kg/day, increased by 20 mg/kg/day every 2 weeks to reach a maximum dose of 40 to 60 mg/kg/day given twice a day. For children younger than 4 years, dose is determined by the doctor.
2. Formulation. Tablets: 250 mg, 500 mg, 750 mg, 1,000 mg; oral solution, 100 mg/mL.
3. Side effects. Fatigue, coordination problems, sleepiness, mood and behavior changes, such as anger, anxiety, depression, hostility, and irritability.
I. Zonisamide (Zonegran; Eisai).
1. Administration. 4 to 8 mg/kg/day divided twice a day. Therapeutic blood levels are 10 to 30 mg/mL. May be effective in myoclonic seizures as in progressive myoclonic epilepsies such as Unverricht–Lundborg syndrome (Baltic myoclonus).
2. Formulation. Capsule: 25 mg, 50 mg, 100 mg.
3. Side effects. Somnolence, dizziness, anorexia, headaches, confusion, cognitive impairment, oligohidrosis hyperthermia, and renal calculi (4%). Contraindicated in care of patients with hypersensitivity to sulfonamides (zonisamide is a sulfonamide).
J. Felbamate (Felbatol; Wallace).
1. Administration. Begin at 15 mg/kg/day for first week, 30 mg/kg/day for the second week, and 45 mg/kg/day divided three times a day from the third week (maximum 3,600 mg/day). Therapeutic blood levels are 50 to 110 mg/mL.
2. Formulation. Tablets: 400 mg, 600 mg; oral suspension 600 mg/5 mL.
3. Adverse effects. Gastrointestinal side effects, including weight loss and anorexia, have been most prominent. Insomnia, somnolence, and fatigue also have occurred. Rash may occur if the patient is also taking Depakote. On August 1, 1994, a year after felbamate was approved, several cases of aplastic anemia and hepatotoxicity were reported. Physicians should restrict use of this drug only to children with severe epilepsy, especially LGS, which is refractory to other therapies with close monitoring of blood work according to specific guidelines.
K. Ethosuximide (Zarontin; Parke-Davis).
1. Administration. 20 to 40 mg/kg/day in two or three divided doses. Therapeutic blood levels are 40 to 100 mg/mL.
2. Formulation. Capsules: 250 mg; syrup: 250 mg/5 mL
3. Side effects. Gastric irritation, anorexia, nausea, vomiting, drowsiness, and hallucinations
L. Clonazepam (Klonopin; Roche).
1. Administration. 0.03 to 0.10 mg/kg/day in two or three divided doses. Therapeutic blood levels are 0.02 to 0.08 mg/mL.
2. Formulation. Tablets: 0.5 mg, 1 mg, 2 mg
3. Side effects. Drowsiness, ataxia, irritability, diplopia, and drooling
M. Clorazepate (Tranxene; Abbott).
1. Administration. 0.3 mg/kg/day, increased to a maximum of 3 mg/kg/day. Therapeutic blood levels are not established.
2. Formulation. Tablets: 3.75 mg, 7.5 mg, 15 mg
3. Side effects. Drowsiness, dizziness, ataxia, and drooling
N. Rufinamide (Banzel; Eisai).
1. Administration. Therapy should be initiated at 10 mg/kg/day in two divided doses, to be increased by 10 mg/kg increments every 2 days to a target dose of 45 mg/kg/day or 3,200 mg/day whichever is less.
2. Tablets. 200, 400 mg; suspension: 40 mg/mL
3. Side effects. Somnolence, headache, dizziness, fatigue, and somnolence. It is contraindicated in patients with familial short QT syndrome. Multiorgan hypersensitivity syndrome has been reported in association with Banzel therapy, especially in children <12 years of age.
O. Vigabatrin (Sabril; Lundbeck).
1. Administration. Initial dosing of 50 mg/kg/day in two divided doses is increased by 25 to 50 mg/kg/day every 3 days up to a maximum of 150 mg/kg/day.
2. Formulation. 500 mg powder per packet for oral solution; 500 mg tablet
3. Side effects. Progressive and permanent bilateral concentric visual field constriction has been noted in 30% or more patients. It can be mild to severe resulting in disability. It should therefore be withdrawn from a pediatric patient treated for infantile spasms who fails to show benefit within 2 to 4 weeks. Abnormal MRI signals have been noted in some, but they generally resolve with discontinuation of therapy. Anemia, somnolence, and weight gain have also been reported.
P. Clobazam (Onfi; Lundbeck).
1. Administration. Therapy is initiated at a low dose. Dose range is 10 mg to 40 mg/day, and 0.4 to 0.8 mg/kg/day in children <12 years of age, administered in a bid dosing.
2. Formulation. Tablet: 10 mg, 20 mg; suspension: 2.5 mg/mL
3. Side effects. Common side effects include drowsiness and sedation. Dry mouth, and behavioral problems including restlessness and aggressive outbursts have also been noted.
Q. Lacosamide (Vimpat; UCB Pharma).
1. Administration. Approved for adults with partial-onset seizures. Start with 50 mg/day increasing to a maintenance of 200 to 400 mg/day. Lower doses are used in children.
2. Formulation. Tablet: 50 mg, 100 mg, 150 mg, 200 mg; oral solution 10 mg/mL; IV solution: 200 mg/20 mL.
3. Side effects. Prolonged PR interval, multiorgan hypersensitivity reaction, dizziness, ataxia, fatigue, nausea, blurred vision, tremor
R. Perampanel (Fycompa).
1. Administration. Approved for treatment of partial-onset seizures in patients ≥12 years of age. Start at 2 mg at night increasing to maintenance of 8 to 12 mg/day. If added to enzyme-inducing AEDs such as PHT, CBZ, and OXC, start at 4 mg at night.
2. Formulation. Tablets: 2 mg, 4 mg, 6 mg, 8 mg, 10 mg, 12 mg
3. Side effects. Anger, aggression, irritability, dizziness, somnolence. Violent thoughts or threatening behavior including homicidal ideation has been observed in a few patients.
S. Other antiepileptic medications available in the United States include tiagabine (Gabitril; Abbott), gabapentin (Neurontin; Parke-Davis), and pregabalin (Lyrica; Parke-Davis). Stiripentol used for Dravet’s syndrome is currently not approved in the United States.
PSYCHOSOCIAL ISSUES
A. Avoid risk factors such as fatigue and sleep deprivation. Seat belts and bicycle helmets should be worn to prevent head injuries that may lead to seizures.
B. Seizure precautions. Bathtubs should be avoided; only showers should be taken. Activities such as climbing of heights, swimming without supervision, driving, contact with heavy machinery and fire, and other activities that could be dangerous in the event of a seizure should be avoided.
C. Parents should guard against overprotection, which can develop out of fear and anxiety. Unnecessary limitations prevent the child from taking the risks necessary for him or her to become an independent person and develop self-confidence.
D. Parents must inform schoolteachers (as well as babysitters) about the child’s seizures. This allows teachers to be prepared to deal with seizures in the classroom and the reactions of classmates. Teachers should be expected to provide information about frequency of seizures during school hours, changes in the child’s behavior, unexplained changes in school performance, and abnormal behavioral and social problems that may require referral for counseling.
E. If certain activities must be restricted because of poor seizure control, substitute exercise programs must be found.
F. A medication schedule that avoids school hours should be planned because it is often inconvenient and embarrassing for a child to take AEDs at school.
G. Children with epilepsy have an increased risk of learning disabilities, attention-deficit hyperactivity disorder, anxiety, and depression. The Food and Drug Administration (FDA) has warned that AEDs may be associated with suicidal ideation.
H. Referral to services such as the Epilepsy Foundation of America or local support groups for counseling.
STATUS EPILEPTICUS
DEFINITION
One or more seizures lasting for more than 30 minutes without full recovery of consciousness between seizures
A. Generalized convulsive status epilepticus is characterized by persistent GTCS. In children, it is associated with a higher morbidity and mortality than in adults. We, therefore, focus on the management of this type of status epilepticus.
B. Nonconvulsive status epilepticus includes cases of absence status and complex partial status and is often described as “twilight state.”
PRECIPITATING FACTORS
A. Abrupt discontinuation, noncompliance, or changes in anticonvulsant therapy
B. Acute intercurrent infections such as meningitis and encephalitis
C. Acute metabolic disturbances, such as electrolyte disturbances and hypoglycemia
D. Acute cerebral insult such as anoxia, hypoxia–ischemia, trauma (SAH, subdural hematoma, and depressed skull fractures)
PROGNOSIS
Approximately 15% of all patients with epilepsy have an episode of status epilepticus at some time in their lives. It is more common in those younger than 2 years of age. Recurrent status epilepticus is more common among children. In the pediatric age group, the mortality rate is approximately 3% to 11%, with higher rates within the first 6 months of life.
TREATMENT
A. Confirmation of diagnosis of status epilepticus. The longer the seizure continues, the more difficult it is to control and the greater the possibility of permanent brain damage.
B. General measures.
1. Initiate supportive measures—see Table 42.4.
2. Place an IV line (preferably two, one with normal saline solution). Blood should be obtained at this time for CBC, electrolytes, calcium, magnesium, phosphorus, glucose, liver function tests, AED levels, toxicology screening, and blood cultures if febrile.
3. Administer IV 25% glucose at 2 mL/kg.
4. Identify and treat precipitating factors. For a patient with known seizures in whom status epilepticus may have been caused by AED withdrawal, the treatment of choice is reinstatement of the same drug.
C. Drug treatment involves administration of a drug for immediate termination of the seizure and a second drug for maintenance therapy. The initial treatment is similar regardless of the type of seizure. Maintenance treatment varies depending on the type of epilepsy. The protocol is presented in Table 42.4.
1. Benzodiazepines.
a. Lorazepam (Ativan; Wyeth-Ayerst) is recommended as a first-line treatment. The advantages are rapid onset of action, prolonged antiepileptic activity compared with diazepam, less respiratory depression and sedation, and a lower rate of seizure recurrence. IV diazepam (0.2 to 0.5 mg/kg/dose, maximum of 5 mg administered over 2 to 5 minutes) is not routinely used because of disadvantages including short duration of action (<30 minutes), high incidence of respiratory depression, and tendency to precipitate tonic status in patients with LGS.
2. Fosphenytoin (Cerebyx; Parke-Davis). After IV lorazepam has been administered, IV fosphenytoin is given to achieve therapeutic levels of 18 to 20 mg/mL. Fosphenytoin is a water-soluble disodium ester prescribed as equimolar amounts of phenytoin called PEs. This prodrug is rapidly converted to phenytoin by phosphatase in the blood stream, reaching peak brain levels 15 minutes after administration. The loading and maintenance doses of fosphenytoin in PEs are identical to those of phenytoin. Fosphenytoin can be administered IV and intramuscularly with minimal local tissue damage and at faster rates of administration with fewer adverse effects than with phenytoin. Pruritus and paresthesia can occur. IV phenytoin can also be used. Disadvantages of phenytoin include cardiac arrhythmias and hypotension requiring close electrocardiographic and blood pressure monitoring. Intramuscular administration is avoided because of unpredictable absorption and muscle irritation. IV extravasation can result in phlebitis and tissue necrosis. Poor absorption occurs in children, resulting in difficulty in maintaining steady therapeutic levels, especially when the patient is switched to the oral form for maintenance. Phenytoin should be administered in normal saline solution, because it precipitates in glucose solutions.

3. IV phenobarbital is administered if seizures persist. It is often used as the first choice in children under 6 years of age. In the treatment of neonates, this may be administered as a single dose. In the case of older children, it may be divided into aliquots of 10 mg/kg to avoid respiratory depression until seizures stop or a maximum loading dose of 20 mg/kg. Additional 10 to 20 mg/kg may be necessary to achieve therapeutic levels of 35 to 40 mg/mL. Disadvantages of phenobarbital include hypotension and respiratory depression. IV valproate and levetiracetam are not currently FDA approved for treatment of status epilepticus.
4. A more detailed history interview and neurologic examination should be performed at this time. Evaluate the initial blood work. Before initiating additional therapies, it is preferable to obtain CT scans without contrast enhancement and to perform LP to look for causes such as intracranial structural lesions or infections. Consider LP for any child with a fever, especially if younger than 18 months, because meningitis can occur without clinical signs of neck stiffness.
5. Refractory status epilepticus. If seizures persist for 60 minutes and the patient does not respond to loading doses of fosphenytoin and phenobarbital, anesthetizing agents such as pentobarbital, midazolam, or propafol should be considered. Phenobarbital can be used in additional IV boluses of 5 to 10 mg/kg with EEG monitoring until seizures stop and a burst-suppression pattern is obtained on the EEG. The disadvantage of phenobarbital coma is that because of a longer half-life, the effect takes longer to wear off.
The recommended duration of pharmacologic induced coma is 48 to 72 hours. During this time, the patient is rechecked for seizures by means of decreasing the infusion rate. If seizures persist, the procedure is repeated. If seizures are adequately controlled, medication is slowly withdrawn. Administration of coma requires an intensive care unit setting with controlled mechanical ventilation and close cardiac monitoring.
6. If seizures are still not controlled, general anesthesia with halothane and neuromuscular blockade is recommended.
MEDICATION WITHDRAWAL
Although there is no consensus on how long a patient should remain seizure-free before drug withdrawal is considered, a seizure-free period of 2 to 4 years is recommended. Relapse rates are higher among adults than among children. Approximately 50% of seizure recurrences occur within 6 months of tapering the AED, and 60% to 90% occur within the first year. Children with febrile seizures have a 97% chance of outgrowing them by 6 years of age. There is an 80% to 85% chance of remission among children with only absence seizures.
FAVORABLE FACTORS ASSOCIATED WITH LOWER RELAPSE RATE
A. Reasonable ease of seizure control
B. Normal neurologic examination findings and developmental milestones
C. Normal EEG findings at time of withdrawal
D. Seizure onset between 2 and 12 years of age
E. Certain seizure types—simple febrile seizures, BECTS, childhood absence epilepsy.
UNFAVORABLE FACTORS ASSOCIATED WITH INCREASED CHANCE OF RECURRENCE
RECURRENCE RATE BEING HIGHER IN REMOTE SYMPTOMATIC EPILEPSY (EPILEPSY DUE TO AN IDENTIFIABLE CAUSE)
A. Recurrence rate is higher in remote symptomatic epilepsy (epilepsy due to an identifiable cause)
B. Seizures of long duration before successful establishment of control
C. Abnormal EEG findings at time of medication withdrawal
D. Abnormal neurologic examination findings
E. Seizure onset <2 years of age or after 12 years of age
F. Certain seizure types—focal seizures, infantile spasms, LGS, juvenile myoclonic epilepsy.
VAGAL NERVE STIMULATION
In the United States, FDA approval indicates use of vagal nerve stimulation as adjunctive therapy for refractory partial seizures in the care of adults and children older than 12 years. It has also successfully been used in younger children for other seizure disorders such as LGS. Indications for consideration of the device include medically refractory seizures, adequate trial of at least three AEDs, exclusion of nonepileptic events, and lack of surgery candidacy. Children who have undergone vagotomy (unilateral or bilateral) should not be given implants. Cervical masses should be excluded because cervical MRI may not be performed once the device is implanted. Cardiac arrhythmias, conduction abnormalities, and chronic obstructive pulmonary diseases are also considered as risk factors. Common side effects include voice hoarseness, cough, dyspnea, and rarely left vocal cord paralysis and parasthesias. It is associated with a 50% reduction in seizure activity in 30% to 50% of individuals.
Procedures that resect or disconnect epileptogenic areas can reduce or eliminate seizures in patients with medically intractable epilepsy. These procedures are performed in specialized epilepsy centers. Extensive preoperative evaluation includes video EEG monitoring to identify seizure focus, neuropsychiatric evaluation, neuroimaging studies (MRI, single-photon emission computed tomography, and PET), intracarotid amobarbital test (Wada’s test), and even invasive studies (subdural and depth electrodes), if indicated. The most common types of epilepsy surgery in the care of children are as follows.
RESECTIVE SURGERY
Removal of the epileptogenic area (e.g., temporal lobectomy)
CORPUS CALLOSOTOMY
Interruption of the anterior two-thirds of the corpus callosum, effective for atonic seizures, tonic seizures, and GTCS
HEMISPHERECTOMY
One cerebral hemisphere is disconnected from the rest of the brain, and a limited area is resected. It is performed for early-onset or congenital hemiplegia in which seizures arise from one side of the brain.
Key Points
• Currently, epilepsies are classified as localization-related (focal, partial), generalized epilepsies and epileptic syndromes, epilepsies and syndromes undetermined whether focal or generalized, and situation-related seizures.
• Epileptic encephalopathies often begin early and are characterized by severe cognitive and behavioral impairment and frequently intractable seizures. Examples include Ohtahara’s syndrome, Dravet’s syndrome, MAE, infantile spasms, and LGS.
• Neonatal seizures are more fragmentary than seizures after 2 months of age and have multiple possible causes including hypoxia-ischemia, metabolic derangements, trauma, vascular lesions, infections, CNS anomalies, inherited metabolic or genetic syndromes, and drug withdrawal.
• Febrile seizures are common, affect 2% to 5% of children, have a peak onset of 18 to 24 months, and typically remit by 6 years of age.
• Beyond basic metabolic studies (glucose, calcium, electrolytes), EEG, and neuroimaging, evaluation of seizures in children may require LP for seizures with fever, assessment for inborn errors of metabolism in children with seizures and regression, and chromosomal microarray for seizures in children with dysmorphic features and in children with epileptic encephalopathy.
• First seizures do not require AEDs unless there are abnormalities on the neurologic examination, epileptic discharges on EEG, abnormalities on neuroimaging, or diagnosis of epilepsies with a high recurrence risk such as infantile spasms, myoclonic or atonic epilepsies, or absence epilepsy.
• After 2 to 4 years of seizure freedom, favorable factors for successful withdrawal of AEDs include ease of seizure control, normal neurologic examination, normal EEG at the time of medication withdrawal, seizure onset between 2 and 12 years of age, and specific seizure types such as benign epilepsy with centrotemporal spikes and childhood absence epilepsy.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


