Experimental Therapies for Spinal Cord Injury
Marion Murray
Introduction: The Problems of Spinal Cord Injury
Many factors conspire to prevent repair and recovery after spinal injury. The intact adult spinal cord contains inhibitory molecules that cause collapse of growth cones, which are the elongating tips that begin to emerge from damaged axons, thus preventing regenerative sprouting. The injury itself creates a hostile environment; the entrance of inflammatory cells and release of toxic compounds from injured cells lead to a secondary expansion of the injury. The glial scar, a barrier to regeneration, then develops. The adult central nervous system (CNS) neuron is itself poorly equipped to regenerate. These neurons, unlike peripherally projecting neurons, do not readily express genes needed for axonal elongation and, in addition, they are more vulnerable to axotomy and thus may demyelinate, atrophy, or die when injured. These pathological changes may diminish functional contributions by intact collaterals of damaged neurons even to circuits not directly affected by the injury (Fig. 12-1).
These daunting obstacles are yielding to a series of novel research directions. Many of the barriers to regeneration, whether intrinsic or extrinsic to the neuron, and whether normally present or associated with the injury, can now be at least partially breached. Although treatments directed to individual components have led to only incremental improvements in regeneration and recovery of function, there is hope that appropriate combinations of treatments targeted at mechanistically different impediments will ultimately result in a substantially greater extent of recovery and repair. We are looking not for a magic bullet but perhaps for a magic fusillade.
Experimental Injury Paradigms
The search for the most appropriate animal model for developing and validating spinal cord injury (SCI) treatments has focused on those that more closely mimic injuries seen in the clinic, such as contusion or compression injuries. These are incomplete injuries that often leave the dura intact and spare, to a variable extent, white matter while destroying much of the gray matter at the injury site. Other models include surgical lesions that interrupt specific white matter tracts, or funiculi, and thus permit a more focused mechanistic investigation of the effects of injury and treatment on identified pathways. The type of injury model that is most appropriate is, of course, dependent on the questions being asked, but is
likely to progress from the more analytical approach to the more clinically relevant (1).
likely to progress from the more analytical approach to the more clinically relevant (1).
 FIGURE 12-1. Diagram of spinal cord following a contusion injury. A contusion injury induces hemorrhage and may or may not damage the dura. The epicenter is characterized by necrotic cell death and the lesion expands (secondary injury) into a penumbra region, with additional cell death due to apoptosis. The secondary injury is associated with entry of inflammatory cells and toxicity from dying spinal cord cells. Ultimately, the area of injury is surrounded by a dense glial scar. The injury destroys some axons, and damages others, leading to demyelination. Cut axons often retract (dieback). Some axons are usually spared by a contusion injury. |
The emphasis over the past decade has been on demonstrating efficacy of single treatments as measured with one or more outcome measures. One of the encouraging features of the research being carried out currently is the development and validation of a range of outcome measures that may include axonal growth, neuroprotection, and behavioral measures targeting different functions. Anatomical repair has been assessed by evidence of axonal growth, using tracing methods to estimate increased density or distance of growth; both regeneration and sprouting have shown stimulation through various treatments. Neuroprotection has been evaluated by lesion size to estimate effects on secondary damage, or by cell counts of surviving axotomized neurons to estimate rescue from retrograde degeneration; some treatments have resulted in decreased lesion size and others have shown rescue of neurons distant from but axotomized by the lesion. Functional improvement has usually been evaluated using one or more behavioral measures. Most commonly, tests of overground locomotion have been used, and a variety of treatments have shown improvement in motor function. More recently, in response to the needs articulated by SCI patients, treatments that may ameliorate autonomic disorders (e.g., bladder, bowel, sexual dysfunction, or neuropathic pain) are being emphasized.
There has been no shortage of positive findings from experimental studies. The emphasis in the next decade will be on determining efficacy of individual treatments from different laboratories and in developing combinatorial treatments that target different mechanisms.
New Treatments from the Laboratory
Rescuing Injured Neurons
Necrosis
Necrosis is the form of cell death resulting from external insults, including physical injury, glutamate toxicity, and ischemia, and is the primary cause of cell death following CNS trauma. In CNS injury, the cells in the core of the injury site undergo necrosis (Fig. 12-1), are resistant to treatments, and are probably unsalvageable. The cysts that develop once necrotic tissue is phagocytosed constitute additional barriers to repair.
Apoptosis
Apoptosis is the form of cell death that is amenable to treatment. Neurons in the penumbra, adjacent to the epicenter of the injury (Fig. 12-1), may escape necrosis but remain susceptible to apoptosis. These neurons can be rescued by antiapoptotic treatments, such as inhibitors to caspases, which are enzymes (cysteine proteases) expressed following injury that can kill cells by cleaving critical repair and homeostatic proteins as well as cytoskeletal proteins (2). The effect of diminishing apoptosis would be a decrease in the secondary expansion of the lesion and thus an improvement in the prospects for recovery. In the adult CNS, apoptosis is mediated through pathways both extrinsic and intrinsic to the neuron (3) (Fig. 12-2). Treatments to be administered acutely that will detoxify the environment, and thus diminish apoptotic cell death, are currently under active investigation.
The extrinsic apoptotic process is initiated by binding of ligands (e.g., tumor necrosis factor, TNFa) to death receptors located on the neuronal membrane that mediate apoptosis. Intrinsic apoptosis results from mitochondrial damage triggered by ischemia, stress, excitotoxicity, elevated intracellular calcium (Ca++), and other insults. Both intrinsic and extrinsic apoptotic pathways activate caspase cascades leading to degradation of structural proteins, DNA, and ultimately death. These are parallel and interacting death mechanisms, and their relative importance may differ according to neuronal types (Fig. 12-2). Neurons contain a default apoptotic pathway, normally inhibited by extrinsically supplied trophic factors that bind to trophic factor receptors and block apoptosis; there is also an intracellular pathway that blocks the apoptotic pathway. Apoptosis can be suppressed by using decoy proteins that block death receptors or inhibition of pro-apoptotic proteins by apoptotic brakes (e.g., activated protein C [APC], proteins from the B-cell leukemia/lymphoma-2 [Bcl2], or heat shock protein [HSP] families, as well as enzymes such as heme oxygenase) (3). Apoptosis can also be suppressed by providing therapeutic levels of trophic factors that activate survival pathways (4) or caspase inhibitors that act further downstream. Animal research has therefore identified several promising approaches that result in greater survival of vulnerable injured neurons at the site of injury. Nevertheless, there has not yet been successful translation of these therapies to the clinic. The variety of targets and pathways suggests redundancy and increases the likelihood that combined approaches may be needed.
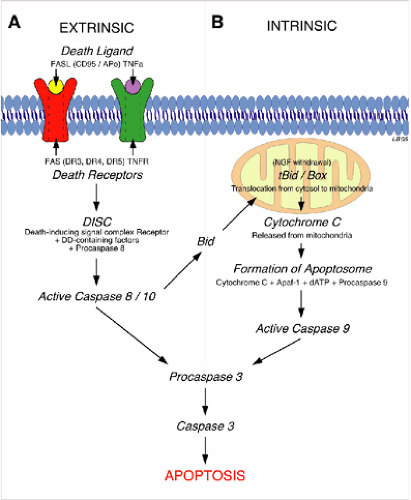 FIGURE 12-2. Extrinsic and intrinsic pathways leading to apoptosis. A: Extrinsic pathways involve binding of a death ligand to death receptors, with subsequent activation of caspases leading to apoptosis. B: The intrinsic pathway is engaged by the activated caspases, leading to apoptosis. The pathways may thus converge within the cell. Identification of the components of the pathways, some of which are indicated here, provides targets for pharmacological intervention. |
Other strategies to diminish secondary injury include suppressing inflammation. In the acute post-injury phase, there is strong up regulation of genes involved in inflammation (5, 6). Although inflammatory responses to SCI clearly contribute to secondary damage, nonselective suppression of inflammation has not proven useful, perhaps because beneficial effects of the inflammatory response are also eliminated. Methylprednisolone, for example, is an anti-inflammatory, antioxidant steroid approved for use in human SCI patients but whose benefits have been questioned (7, 8 and 9). Given the complex cascade of bioactive molecules that are released upon injury, it is not surprising that components of the inflammatory process may promote repair; therefore, the inhibition of inflammatory events might have deleterious consequences. Studies of optic nerve injury have shown that apoptosis is reduced and regeneration is enhanced by several interventions that are mediated by a macrophage-derived factor whose release is stimulated by injury (10). Kipnis et al. (11) have proposed that boosting reactive T cells, normally considered to be deleterious, can confer protective autoimmunity and improve function after spinal cord injury and, indeed, clinical trials are under way to test that hypothesis. A study that attempted to replicate those laboratory findings, however, instead showed that T cells activated by SCI are pathological effector cells that impair spontaneous functional recovery and exacerbate tissue injury
(12). Further, macrophage infiltration and phagocyte-mediated degeneration of myelin following SCI result in exposure of axons to inhibitory molecules that contribute to dieback of injured axons (13). The discovery that cellular and molecular events thought to be inhibitory to repair may also have positive effects is a recurrent theme as research progresses. Thus, more targeted approaches may be needed to focus on specific deleterious effects while preserving beneficial effects of treatments.
(12). Further, macrophage infiltration and phagocyte-mediated degeneration of myelin following SCI result in exposure of axons to inhibitory molecules that contribute to dieback of injured axons (13). The discovery that cellular and molecular events thought to be inhibitory to repair may also have positive effects is a recurrent theme as research progresses. Thus, more targeted approaches may be needed to focus on specific deleterious effects while preserving beneficial effects of treatments.
There are several examples of more targeted approaches. Following injury, integrin molecules on the cell surface of leukocytes bind to endothelial cell adhesion molecules, resulting in infiltration of neutrophils and entry of hematogenous monocyte macrophages at the injury site; this inflammatory response can result in significant secondary tissue damage. Blockade of an integrin receptor, cd11d/cd18, with monoclonal antibodies following SCI diminishes this inflammatory response, and, if this treatment is applied within the first 48 hours after the injury, there is reduced secondary damage, stimulated axonal growth, and improved outcome with decreased autonomic dysreflexia, decreased neuropathic pain, and improved hindlimb function (14). Another target is the blockade of glutamate receptors. Levels of excitatory amino acids such as glutamate rise rapidly to toxic levels after SCI, leading to neuronal death, glial death, and delayed expansion of the injury. The potent and highly specific antagonist 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(f)quinoxaline (NBQX) is a subset of glutamate receptors, the alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptors (15), and, when administered soon after SCI, results in reduced functional impairment on a range of outcome measures and stimulated axonal growth (16, 17), presumably through a protective effect that results in greater sparing of spinal tissue.
Death at a Distance
Axotomized CNS neurons whose cell bodies are located at a distance from the injury may undergo retrograde degenerative changes. These may include moderate or severe atrophy as well as death of the axotomized neuron. There is controversy about whether some neurons actually die or simply atrophy below the level of resolution; nevertheless, it is likely that axotomized neurons that are severely atrophic will not contribute to useful function even if some of their collateral projections to other targets survive. The retrograde degenerative changes appear to be apoptotic in nature and thus amenable to treatment. Disappearance of Clarke or red nucleus neurons following axotomy with spinal hemisection has been prevented by transplanting fetal spinal cord tissue, by cell grafts that produce neurotrophic factors (NTFs), and by direct injection into the injured spinal cord of the antiapoptotic molecule Bcl-2 or NTFs such as brain-derived neurotrophic factor (BDNF) or neurotrophin-3 (NT-3). These treatments rescue most of the neurons that would otherwise disappear as well as prevent much of the atrophy (18). Delivery of large amounts of BDNF directly into the red nucleus can rescue axotomized neurons even after chronic injuries (19). There are thus a number of plausible approaches to decreasing apoptosis, preserving damaged neurons, and decreasing secondary injury; other intracellular pathways that contribute to neuronal death are being examined and many have shown promising results. Comparative tests will be needed to determine which are the more effective. What is not known is whether these rescued neurons remain functional. This will probably require electrophysiological analyses, which have not yet been done. An important corollary of this is the observation that despite the very promising preclinical work, clinical trials with antiapoptotic molecular approaches have not proven successful.
Countering the Hostile Environment
The pioneering work from the Schwab laboratory focused attention on the inhibitory nature of CNS myelin and its role in blocking axonal outgrowth (20, 21 and 22). The inhibitory molecules associated with CNS myelin are those encountered initially by a regenerative sprout and are primarily responsible for the abortive nature of regenerative sprouting. Huang et al. (23) blocked myelin-associated inhibitors by injecting a vaccine against myelin to stimulate the animal’s own immune system to produce antibodies to these inhibitors. These mice showed dramatic regeneration of corticospinal axons after a hemisection with recovery of hindlimb function. Over the past few years, the mechanisms by which myelin inhibition acts to prevent regenerative growth have begun to be understood. Three inhibitors associated with myelin (NogoA, myelin-associated protein [MAG], and oligodendrocyte myelin glycoprotein [OMgp]) have been identified. These are structurally different molecules but surprisingly all bind to the same receptor, NgR, which is located on the neuronal membrane and results in inhibition of axonal regeneration. The NgR itself requires transmembrane coreceptors, one of which is the p75 receptor (24). Activation of the p75 receptor leads to downstream activation of the GTPase RhoA, and its target Rho dependent kinase (ROCK) (Fig. 12-3). This Rho-ROCK intracellular pathway acts on the intracellular cytoskeletal proteins actin and myosin, resulting in collapse of the growth cone. The importance of this line of research is in the identification of additional targets for pharmacological intervention. NogoA can be blocked by antibodies (e.g., NEP 1-40), and this leads to regenerative growth and recovery of function (25).
The injured CNS is even more inhibitory to axonal growth than the intact CNS. A contrary perspective on the
role of myelin in prevention of axonal growth is offered by Raisman (26). He distinguishes between the observed collapse of the growth cone in culture and the possibility that myelin-based inhibition in vivo may be a mechanism that guides, and thus facilitates, axonal growth through the highly structured environment of the developing CNS. He suggests that reactive astrocytes are the greater villain among the environmental inhibitors. Astrocytes become activated in response to inflammatory cytokines, begin to secrete inhibitory proteoglycans, for example, chondroitin sulphate proteoglycan (CSPG), and establish a dense glial scar (27). These proteoglycans are inhibitory to axon outgrowth in the uninjured adult CNS and a major inhibitory contributor to the glial scar. The scar, which develops within a week or two following injury, thus provides both biochemical and mechanical barriers to injured axons. In the past few years, we have increased our understanding of these inhibitory events, and therapies that diminish their inhibitory activity are being developed (28). CSPG is normally present in the CNS, however, it is produced in greater quantities by reactive astrocytes. CSPG acts through unknown receptors on the neuron to activate the protein kinase C (PKC) pathway; blockade of PKC activity by intrathecal administration of a specific inhibitor, Go6976, will promote regeneration (29). CSPG thus evokes inhibition of axonal growth using a signaling pathway that is different from that employed by myelin inhibitors. It does not produce generalized
growth cone collapse, as does RhoA, but instead retraction of those short processes on the growth cone that contact the CSPG (30, 31). CSPG can be degraded by chondroitinase ABC (ChABC), an enzyme that partially digests the molecule and thus renders it less inhibitory. Delivery of ChABC into an injured spinal cord has been shown to elicit regeneration of corticospinal axons and to support functional recovery (32).
role of myelin in prevention of axonal growth is offered by Raisman (26). He distinguishes between the observed collapse of the growth cone in culture and the possibility that myelin-based inhibition in vivo may be a mechanism that guides, and thus facilitates, axonal growth through the highly structured environment of the developing CNS. He suggests that reactive astrocytes are the greater villain among the environmental inhibitors. Astrocytes become activated in response to inflammatory cytokines, begin to secrete inhibitory proteoglycans, for example, chondroitin sulphate proteoglycan (CSPG), and establish a dense glial scar (27). These proteoglycans are inhibitory to axon outgrowth in the uninjured adult CNS and a major inhibitory contributor to the glial scar. The scar, which develops within a week or two following injury, thus provides both biochemical and mechanical barriers to injured axons. In the past few years, we have increased our understanding of these inhibitory events, and therapies that diminish their inhibitory activity are being developed (28). CSPG is normally present in the CNS, however, it is produced in greater quantities by reactive astrocytes. CSPG acts through unknown receptors on the neuron to activate the protein kinase C (PKC) pathway; blockade of PKC activity by intrathecal administration of a specific inhibitor, Go6976, will promote regeneration (29). CSPG thus evokes inhibition of axonal growth using a signaling pathway that is different from that employed by myelin inhibitors. It does not produce generalized
growth cone collapse, as does RhoA, but instead retraction of those short processes on the growth cone that contact the CSPG (30, 31). CSPG can be degraded by chondroitinase ABC (ChABC), an enzyme that partially digests the molecule and thus renders it less inhibitory. Delivery of ChABC into an injured spinal cord has been shown to elicit regeneration of corticospinal axons and to support functional recovery (32).
The glial scar also has beneficial functions. Reactive astrocytes contribute to repair of the blood-brain barrier, reduce the inflammatory response, limit cellular degeneration, and thus protect healthy tissue (33, 34). The search, therefore, is on to identify ways of preserving the beneficial functions of reactive astrocytes by targeting more specific contributions to their role in blocking axonal growth.
Given the many contributors to the nonpermissive environment of the adult CNS, it is not surprising that blocking only one set of inhibitors has but modest effects on repair or recovery of function. As the range of inhibitors is being identified and ways of countering some of them are being developed, we can expect combinatorial pharmacological approaches to be useful in diminishing the influence of these inhibitory molecules in the injured spinal cord and thus permitting axonal growth.
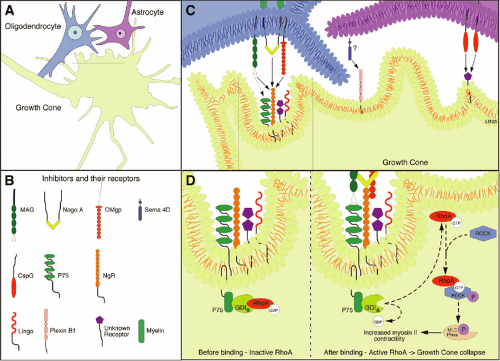 FIGURE 12-3. Inhibitory molecules associated with oligodendroglia and astrocytes produce growth cone collapse. A: Following injury, the growth cone must extend into an inhibitory environment provided by oligodendrocytes and astrocytes. B: Several inhibitors associated with oligodendrocytes and astrocytes have been identified and C: receptors for these molecules have been localized to the growth cone membrane. It is still unclear, however, whether the Semaphorin (Sema 4D) on the astrocyte membrane (indicated by question mark) links to the Plexin B1 receptor at the growth cone. D: Binding to the receptors actives the RhoA pathway, which leads to growth cone collapse and failure of regenerative sprouting. Blockade of receptors for the glial cell inhibitors, or of the RhoA pathway, is associated with increased regeneration and recovery of function. |
Energizing the Neuron
Adult CNS neurons are poorly equipped to regenerate an axon. The growth program that guides developing axons is difficult to access in the adult. Modifying the intrinsic growth potential of the neuron is an important component of a strategy to improve repair. Certain proteins expressed by genes associated with axon regeneration, such as growth-associated protein-43 (GAP-43), tubulin, and membrane proteins, are up regulated by successfully regenerating peripheral nervous system (PNS) neurons. These genes show little response to axotomy in CNS neurons. In the PNS, Schwann cells provide trophic factors that nurture the regenerating PNS neuron and re-establish the genetic program for axonal growth. An inadequate supply of trophic factors in the adult CNS may be a major cause of the difference in regenerative capability between PNS and CNS neurons. The neurotrophins bind to their receptors in the CNS through Trk receptors and an essential coreceptor, p75. Interestingly, p75 also activates the RhoA (35) and the Nogo receptors (36), pathways that cause growth cone collapse. This shared requirement for the coreceptor suggests a competition for access to the coreceptor by neurotrophins that may induce regeneration and inhibitory factors that prevent it; this competition may further limit CNS regeneration. Increasing the supply of neurotrophins to the CNS can improve axonal outgrowth. Indeed provision of trophic factors to the injured spinal cord, either exogenously or through transplantation of cells engineered to secrete trophic factors, is adequate to elicit modest growth of CNS axons, accompanied in some cases by recovery of function (37, 38, 39, 40 and 41). There remain questions of duration and amounts of neurotrophins needed. Treatments that include exogenous administration of large quantities of neurotrophins appear to be more successful in eliciting regenerative growth, but controlling the duration of NTF secretion may also be important. It is likely that repair processes in the injured CNS may occur within a limited window of time. Thus it may be necessary to develop methods of turning off the expression of the neurotrophins. These methods are being developed for cells genetically modified to produce neurotrophins using regulatory elements to control the expression of the transgene.
Successful axonal regeneration mediated by trophic factors binding to the appropriate receptors on the injured neuron is known to elicit an up regulation of a specific set of regeneration associated genes, including transcription factors, GAP-43, and cytoskeletal proteins. Although it is intuitively obvious that coordinated changes in expression of a great many genes at different times postinjury would be required for efficient regrowth of an axon, the likelihood is that the entire pattern need not be reproduced (42). Still the pattern and nature of gene expression is only poorly understood at present. This will certainly change with the advent of microarray technology that will eventually allow the discovery of the genetic requirements (5, 43, 44 and 45) and, with the development of molecular methods to regulate the expression of genes introduced into the damaged CNS.
A conditioning lesion, in which a sensory axon is injured and then later a second injury is made proximal to the first, is a well-established method to increase regenerative growth by dorsal root axons. Interestingly, this can be mimicked by increasing the intracellular concentration of the cyclic Adenosine MonoPhosphate (cAMP) (46, 47). The cAMP is a second messenger that couples extracellular signaling to changes in gene expression that contribute to neuronal plasticity and acts to promote neurite growth. Normally levels of intracellular cAMP expression in neurons are high during development and then diminish once developmental growth has ceased (48). The cAMP pathway, in fact, is one of the targets of neurotrophins. Increasing cAMP levels in neuronal cell bodies after axotomy in the adult has been shown to elicit a partial regenerative response by increasing expression of components of the axonal cytoskeleton (e.g., alpha-I and beta-III tubulin isotypes) (49). Increasing cAMP in the cell body or providing trophic factors that engage the cAMP pathway can counter the inhibition mediated by the inhospitable CNS environment, and thus shift the metabolic balance of an injured neuron toward axonal growth (47, 50). More practically, systemic injection of rolipram (AG Scientific), an inhibitor of the enzyme that degrades cAMP, has been a successful strategy for promoting
regeneration and recovery of function. When rolipram is administered after SCI, together with a transplant of fetal spinal cord (51), Schwann cells or olfactory ensheathing cells (52), or NT-3 (53), regeneration and, in some cases, recovery of function is demonstrated.
regeneration and recovery of function. When rolipram is administered after SCI, together with a transplant of fetal spinal cord (51), Schwann cells or olfactory ensheathing cells (52), or NT-3 (53), regeneration and, in some cases, recovery of function is demonstrated.
We know that axotomized CNS neurons normally elaborate only feeble growth cones that soon collapse. Although the growth cone is specialized for elongation, the collapse occurs when the inadequate growth cone meets the hostile CNS environment. The collapse of the growth cone is driven by activation of the Rho-ROCK pathway, which regulates actin filament polymerization, assembly, and disassembly and the interaction of actin with myosin (54). The collapse can be prevented by blocking the Rho-ROCK pathway. The potential for developing pharmacological treatments is being eagerly explored. RhoA, a small GTPase molecule, can be blocked by a permeable C3 transferase, and ROCK can be blocked by another small molecule, Y27632. Inhibition of this pathway by either means has been associated with axonal growth and with recovery of function (50, 55, 56). The functional improvements occur early, which suggests that these treatments may also provide some degree of neuroprotection (Fig. 12-3).
Related posts:
 Pathophysiology of Spinal Cord Injury
Plain Film Radiography and Computed Tomography of the Cervical Spine: Part I. Normal Anatomy and Spinal Injury Identification
Imaging of Thoracolumbar Spinal Injury
Plain Film Radiography and Computed Tomography of the Cervical Spine: Part II. Classification and Subtypes of Spinal Injury
Magnetic Resonance Imaging of Acute Spinal Trauma
Postoperative Spinal Imaging
Pathophysiology of Spinal Cord Injury
Plain Film Radiography and Computed Tomography of the Cervical Spine: Part I. Normal Anatomy and Spinal Injury Identification
Imaging of Thoracolumbar Spinal Injury
Plain Film Radiography and Computed Tomography of the Cervical Spine: Part II. Classification and Subtypes of Spinal Injury
Magnetic Resonance Imaging of Acute Spinal Trauma
Postoperative Spinal Imaging
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


