Familial Adult Myoclonic Epilepsy (FAME)
Eiichiro Uyama*
Ying-Hui Fu†
Louis J. Ptácek†,‡
*Department of Neurology, Kumamoto University School of Medicine, Kumamoto, Japan
†Department of Neurology, University of California San Francisco, San Francisco, California
‡Howard Hughes Medical Institute, San Francisco, California
Introduction
The identification and characterization of genes responsible for monogenic inherited epilepsies is an exciting area of epilepsy research (1,2,3,4). It is critical for understanding the molecular basis of neuronal hyperexcitability, the etiology of epilepsy, and myoclonic jerks, and, ultimately, the development of new therapeutic agents (3). Ion channels are critical for the regulation of excitability in the central nervous system. In fact, mutations in ion channel encoding genes have been identified in a variety of epileptic disorders (2,3,4).
Several epilepsy disorders are characterized by combinations of symptoms such as focal or generalized and myoclonic seizures (1,2). These disorders include severe myoclonic epilepsy of infancy (SMEI) (2), progressive myoclonic epilepsy (PME) (5), juvenile myoclonic epilepsy (JME) (6), and familial adult myoclonic epilepsy (FAME) (7,8,9). FAME is clinically characterized by autosomal dominant inheritance, adult-onset cortical myoclonic tremor, and infrequent epileptic seizures despite constant paroxysmal discharges on electroencephalogram (EEG). JME differs from FAME with regard to age of onset (early adolescent), genetic transmission (complex), and prevalence of myoclonic jerks upon awakening.
Myoclonic epilepsy is a defining symptom in all PME syndromes, in which myoclonic seizures seem to be generated secondarily due to neuronal degeneration rather than being a primary disorder of neuronal excitability. In contrast, myoclonic jerks in patients with JME or FAME seem to be generated from a functional abnormality of membrane excitability in neurons. In comparison with JME that is a common form of epilepsy, FAME is thought of as a rare form restricted to Japan. However, non-Japanese FAME or FAME-like families have been recently discovered around the world (10,11,12,13,14,15,16) and data now supports locus heterogeneity for FAME.
History
In 1984, Uyama examined the proband in Family 1 (Fig. 22-1) who showed postural finger tremulous movement mixed with myoclonic jerks from the age of 40. She had two episodes of epileptic seizures in her life. The family, originating in Kumamoto in southern Japan, included ten other patients through three generations.
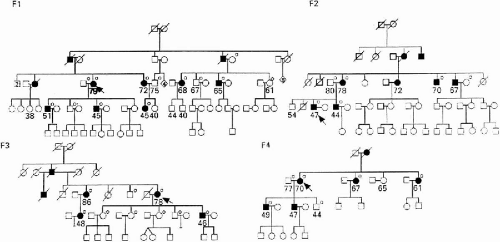 FIG. 22-1. The pedigrees of four FAME families in Kumamoto. Numbers under symbols are ages at examination. An arrow indicates the proband. The filled symbols are affected individuals. Persons with a line over their symbols are deceased. Small circles in the right upper side are subjects studied with DNA analyses. |
Their interictal EEG always displayed epileptic discharges and cortical reflex myoclonus was implicated on somatosensory-evoked potentials (SEP). None of them revealed mental deterioration, cerebellar ataxia, progressive course, or abnormal findings on brain computed tomography (CT)/magnetic resonance imaging (MRI). Because of its unrecognized phenotype at that time, Uyama reported the details at the 26th Annual Meeting of Japanese Neurological Society in 1985 (7). By 1990, another three unrelated families were identified (Fig. 22-1).
Independently, Inazuki et al. (17) collected 12 similar families including 40 patients from Niigata in northern Japan. Based on clinicopathologic features, they proposed the new entity “familial essential myoclonus and epilepsy (FEME)” in a Japanese journal. Coincidentally, Ikeda et al. (18) found two familial cases in Kyoto who had similar features to Family 1. By electrophysiologic analysis, they clarified the nature of tremulous movement and defined it as “cortical tremor.” Later, they presented similar families as “familial cortical myoclonic tremor (FCMT)” (19).
In another independent study, Yasuda found two Japanese families in Okayama that also shared similar features to those in Family 1. Based on electrophysiologic studies and incomplete review, excluding the report of Family 1, and one decisive articles (20), he proposed another term, “benign adult familial myoclonic epilepsy (BAFME)” instead of FEME (22). One of these BAFME families had been described as progressive myoclonic epilepsy (PME) in 1983 (23). Kuwano et al. (24) verified that four Kumamoto families and one BAFME family in Okayama had neither DRPLA mutation nor linkage with γ-aminobutyric acid (GABA) receptor subunits, GABARβ1, GABARβ3, and GABARα6.
Regarding long-term follow-up, Uyama found that the degree of myoclonic jerks in patients increased with age but there was no degenerative components to the phenotype. However the symptoms did not resolve completely as in other benign epilepsies. He then presented the details of four families at the 121st Annual Meeting of ANA in 1996 to be established as a new phenotype of autosomal dominant myoclonic epilepsy of “familial adult myoclonic epilepsy (FAME)” (8). In 1996, efforts to map the FAME gene began with linkage analysis and the FAME locus on chromosome 8q was identified (9). In 1997, Okuma et al. (25,26) reported a similar small family as “familial cortical tremor with epilepsy (FCTE) (Table 22-1).”
TABLE 22-1. Myoclonic Epilepsy Gene Loci with Genes Identified (July 2003) | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
Related posts:
 Progressive Myoclonus Epilepsies: EPM1, EPM2A, EPM2B
Progressive Myoclonus Epilepsies: EPM1, EPM2A, EPM2B
 Myoclonic Status in Nonprogressive Encephalopathies
Myoclonic Status in Nonprogressive Encephalopathies
 Myoclonic–Astatic Epilepsy of Early Childhood – Definition, Course, Nosography, and Genetics
Genetics of Juvenile Myoclonic Epilepsy: Faulty Components and Faulty Wiring?
Ketogenic Diet in Patients with Dravet Syndrome and Myoclonic Epilepsies in Infancy and Early Childhood
Myoclonic–Astatic Epilepsy of Early Childhood – Definition, Course, Nosography, and Genetics
Genetics of Juvenile Myoclonic Epilepsy: Faulty Components and Faulty Wiring?
Ketogenic Diet in Patients with Dravet Syndrome and Myoclonic Epilepsies in Infancy and Early Childhood
 Treatment of Myoclonic Epilepsies of Childhood, Adolescence, and Adulthood
Treatment of Myoclonic Epilepsies of Childhood, Adolescence, and Adulthood
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


