Figure 29-1 T1-weighted sagittal magnetic resonance imaging (MRI) scan of the cranicervical junction (left) showing marked atrophy of the entire spinal cord without associated signal abnormality, and T2-weighted sagittal MRI scan of the cervical and thoracic spine (right).
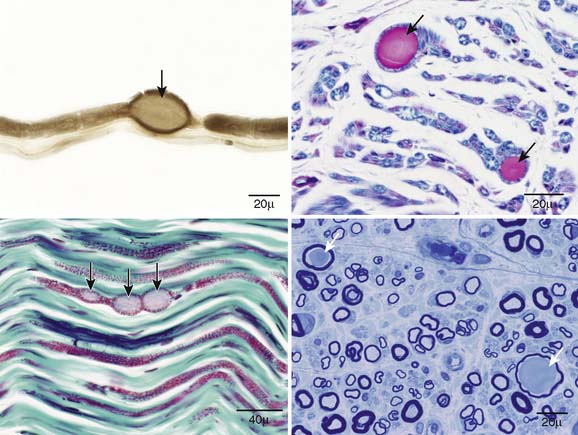
Figure 29-2 Sural nerve biopsy. Teased fiber preparation (top left), luxol fast blue and periodic acid–Schiff preparation (top right), trichrome preparation (bottom left), and epoxy-embedded semithin sections (bottom right) with characteristic polyglucosan bodies (arrows).
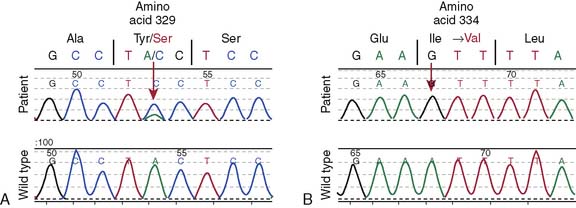
Figure 29-3 Electropherogram showing (A) the Tyr329Ser heterozygous change, which when found in homozygous state, is the most common genetic cause of adult polyglucosan body disease among Ashkenazi Jewish persons, and (B) the homozygous polymorphism Ile334Val found also in unaffected populations (rs2172397). Whether the combination of these two GBE1 DNA changes is disease causing remains undetermined.
CONCLUSIONS
APBD has to be considered in the differential diagnosis of myeloneuropathy, particularly in patients of Ashkenazi-Jewish origin. Not all patients with APBD have reduced GBE activity in available assays. Although a specific homozygous missense mutation in the GBE1 gene is the most frequently reported mutation in this disorder, manifesting carriers of that mutation, other mutations of the GBE1 gene, and individuals without identified mutations in GBE1 have been reported.1,3,9–11 Sural nerve biopsy or skin biopsy from the axilla can provide pathologic confirmation of the disorder and prove particularly helpful in patients with normal GBE activity. On the other hand, rare, small polyglucosan bodies in nerve specimens must not be overinterpreted as evidence of APBD, because they may occur as nonspecific findings in nerve biopsies of older individuals.
Related posts:
 Malignant Peripheral Nerve Sheath Tumor
Disseminated Sporotrichosis with Multiple Granulomatous Mononeuropathies
Late Sporadic CMT4C—A New KIAA1985 Mutation
Late-Onset Transthyretin Val30Met Familial Amyloid Polyneuropathy Unrelated to Endemic Foci
A Weak, and Numb Patient with Tremor—Antimyelin-Associated Glycoprotein Polyneuropathy
Length-Related Axonal Loss in Neuropathy
Malignant Peripheral Nerve Sheath Tumor
Disseminated Sporotrichosis with Multiple Granulomatous Mononeuropathies
Late Sporadic CMT4C—A New KIAA1985 Mutation
Late-Onset Transthyretin Val30Met Familial Amyloid Polyneuropathy Unrelated to Endemic Foci
A Weak, and Numb Patient with Tremor—Antimyelin-Associated Glycoprotein Polyneuropathy
Length-Related Axonal Loss in Neuropathy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


