Introduction
The generalized tonic–clonic seizure (GTCS) has been known since antiquity. The earliest descriptions appear in Egyptian hieroglyphics before 700 B.C.99 From the mid-19th century, when notions of cerebrovisceral sympathy were discarded and all epilepsy was recognized as originating in the brain, the GTCS was considered the cardinal manifestation of genuine or idiopathic epilepsy due to a predisposition to have seizures. GTCS was described as grand mal,49 a term that is still encountered. This predisposition was, in turn, believed to be associated only with generalized seizures (reviewed in Gastaut42). Although the history of clinical epileptology has been marked by the differentiation of nonconvulsive seizures as epileptic events, progressively more exact descriptions of seizure types and epileptic syndromes, the development of surgery (usually for intractable partial seizures), and progress in childhood seizure disorders, the GTCS is still the hallmark of epilepsy for the general public. It remains a dramatic and often frightening event for patients, families, and onlookers. For most neurologists, however, the generalized convulsion is a rarely witnessed clinical event, albeit a common reason for consultation the history of which can be frustratingly difficult to obtain. It may be an isolated event in the setting of an acute encephalopathy, a symptom of serious brain disease, or a manifestation of several epilepsy syndromes of generally good prognosis.
Definitions
The current International Classification of Epileptic Seizures23,30 defines GTCS within the group of generalized convulsive or nonconvulsive seizures. Unlike the conceptual approach of the classification of epileptic syndromes,24 this classification of seizures is based on observed events, specifically, the seizures themselves as documented during intensive monitoring, and the electroencephalogram (EEG). As a group, in generalized seizures, “The first clinical changes indicate initial involvement of both hemispheres…. Motor manifestations are bilateral. The ictal electroencephalographic patterns initially are bilateral, and presumably reflect neuronal discharge which is widespread in both hemispheres.” The classification, however, treats the term tonic–clonic seizures as self-explanatory. In the last proposal by the International League Against Epilepsy (ILAE) Task Force,33 the Glossary of descriptive terminology for ictal semiology describes a tonic–clonic seizure as “a sequence consisting of a tonic followed by a clonic phase. Variants such as clonic–tonic–clonic may be seen.” Dreifuss29 provides a useful description:
“The majority lose consciousness without any premonitory symptoms. There is a sudden sharp tonic contraction of muscles; when this involves respiratory muscles, there is a cry or moan. The patient falls to the ground …tonic contraction inhibits respiration.… The tongue may be bitten.… This tonic stage then gives way to clonic convulsive movements that last for a variable period of time.”
Cyanosis, salivation, tongue biting, and incontinence are frequent, and the postictal state includes a variable period of unconsciousness.
There is at present no provision in the international classification for subclassifying GTCS with preceding bilateral myoclonic jerks or absence attacks. Sequences such as a brief clonic attack before the tonic phase (clonic–tonic–clonic seizure) are also well known to epileptologists, and the glossary indicates this possibility. The classification makes it clear, however, that the term generalized convulsive seizure is restricted to convulsive seizures that are generalized from the start and excludes partial seizures that may become secondarily generalized so quickly as to look like GTCS, as is seen in some frontal lobe seizures.93 Generalized tonic–clonic (convulsive) seizures are manifestations of idiopathic generalized epilepsies (IGEs) and epilepsy syndromes, as these have been defined and, in the most recent proposed classification of epileptic syndromes,33 would be associated with the generalized epilepsies with variable phenotypes: These are discussed in detail elsewhere in this book.
Seizures that often differ from classic tonic–clonic convulsions can occur when partial seizures become generalized. Distinguishing these from GTCS due to an IGE syndrome is a common problem of vital importance in daily practice. Especially in seizures arising in the temporal lobe, clinical spread is not immediate, and the complete seizure pattern (see “Diagnosis,” below) may aid in diagnosis. These seizures are classified under partial seizures as partial seizures evolving to GTCS, although the seizure pattern is often different from that of GTCS. An isolated seizure or brief flurry of seizures clinically indistinguishable from GTCS may occur with an acute diffuse encephalopathy. These are classified as situation-related seizures in the classification of epilepsies, because they do not constitute a separate seizure type. Childhood febrile convulsions, probably the most common form of convulsion, are similarly classified.
It is important to emphasize that, although generalized convulsions may occur in the generalized cryptogenic and symptomatic epilepsies and syndromes, they are not the predominant or defining seizure type and usually happen relatively late in the course of the disease. Generalized seizures manifested only by a tonic phase are, however, quite different from GTCS and are common and often intractable in these syndromes. True GTCSs are not frequent in young children but can occur, particularly in epilepsy with myoclonic–astatic seizures,27 severe myoclonic epilepsy of infancy (Dravet syndrome),28 and in some progressive myoclonic epilepsy varieties in children and adults, such as Jansky-Bielschowsky disease94 and Unverricht-Lündborg disease.68
Epidemiology
Tonic–clonic seizures may be the most common afebrile seizure type in the general population. However, until recently, epidemiologic studies included patients with clinically generalized seizures of partial onset and did not specify the epilepsy syndromes in which GTCSs occur. The occurrence of GTCS is also age-related. Although epilepsy begins much more commonly in infants, children, and the elderly than in middle life, other seizure types are more common in young children, related in part to incomplete synaptic development and myelination, especially of interhemispheric connections.2 Generalized convulsive seizures are uncommon in infants and rarely, if ever, occur in neonates. In the elderly, seizures usually reflect localized brain lesions. Thus, GTCSs are rare in the first few years of life, although myoclonic and tonic seizures are not, whereas partial seizures with secondary generalization are common in the elderly. Typical GTCSs are common, however, in later childhood, adolescence, and young adult life, with the onset of IGEs. Studies based on seizure descriptions without EEG or imaging results or which refer to GTCS in very young children will, thus, overestimate their occurrence. Studies from specialized epilepsy centers that concentrate on patients with intractable seizures and candidates for epilepsy surgery risk underestimating the prevalence of GTCS. It should also be noted that not every subject with a generalized epilepsy syndrome has GTCSs.
Hauser and Kurland,56 using the unique medical records system in Rochester, Minnesota, reported that the prevalence of patients with only GTCS was 20.6% of patients with epilepsy. Patients with GTCS also accounted for 23%54 of the incidence of epilepsy. Incidence estimates for all recurrent afebrile seizures in children and adolescents are roughly similar in different studies, from 50 to 100 new cases per 100,000 per year.55 A study of clinical and EEG data61 estimated that about 39% of patients with epilepsy have a generalized epilepsy syndrome. As discussed later, GTCSs occur in several of these syndromes. Patients with absences commonly develop GTCS, particularly if absences begin relatively late, in preadolescent years: This occurs in about 35% of patients with absences in general, but in 90% of patients with later-onset absences (reviewed in Aicardi3).
A genetic component is evident in many different types of epilepsy. However, the common epilepsies do not usually behave as simple Mendelian disorders, and several lines of evidence suggest multifactorial inheritance.5 The genetics of juvenile myoclonic epilepsy (JME), in which GTCSs often occur, have been recently reviewed.110 Relatives of patients with idiopathic epilepsy have a higher risk for epilepsy than do those of patients with symptomatic epilepsy.86 Population studies show an increased risk of unprovoked seizures in close relatives of patients with idiopathic age-related epilepsy syndromes of childhood. The relative risk ranges from 2.5 (95% confidence interval [CI] 1.3–4.4] for siblings to 3.4 (95% CI 2.1–5.1) for offspring. However, the risk for offspring of parents with GTCS, about 5%, is not higher than that for offspring of parents with partial seizures, the difference being accounted for by the much higher risk to offspring if a parent has absences.7,86 Twin studies in IGEs, reviewed by Berkovic et al.,12 provide further evidence for a large inherited component and show a high rate of concordance in monozygous twins for specific syndromes, including those with GTCS as a major feature. Other risk factors80,92 include a history of febrile convulsions (odds ratio 13.75, 95% CI 3.49–54): This may indicate a lower convulsive threshold and cannot be taken as the cause of later GTCS. Some familial epilepsies are associated with specific ion channel defects (Chapter 18), but single gene defects are a rare cause of common epilepsies. This information is, however, in keeping with a polygenic inheritance for common epilepsies.
Anatomic Pathways and Pathophysiology
Different forms of convulsive seizures in animals depend on independent anatomic regions and pathways. Models of convulsive seizures using drugs such as bicuculline and pentylenetetrazol, kindling studies, and animals genetically prone to generalized epilepsy at times with reflex triggers, have implicated brainstem structures (reviewed by Browning19 and Moraes82) in the genesis and, clearly, in the modulation of convulsive seizures; including the lateral geniculate body, which produces GTCS when kindled in the cat96; ascending pathways through the mamillary bodies and anterior thalamus; the substantia nigra, including a nigrotectal γ-aminobutyric acid (GABA)ergic projection38; and diffuse increased noradrenergic innervation of the cortex from the locus coeruleus.84 The three principal concepts of generalized epilepsy, thus, each implicate a different mechanism: an abnormal response of hyperexcitable cortex to initially normal thalamic input, a primary subcortical trigger, and abnormal cortical innervation from subcortical structures. Subcortical mechanisms would play at least a modulating role. Any or several of these may operate in the clinically and genetically different syndromes with GTCS in humans (for a review with EEG emphasis, see Binnie14). It should be emphasized that GTCS models in rodents and fowl are clinically quite different from human epilepsy; feline, canine, and primate models may be closer approximations.
Studies in the Senegalese baboon Papio papio, which has both photosensitive and spontaneous generalized motor seizures, suggest that the substantia innominata participates in the mechanism of generalized convulsions by modulating excitability of the hemispheric motor mechanisms. In partial seizures that spread from limbic structures, the claustrum participates in activating the ipsilateral hemispheric motor mechanisms. Bilateral spread of partial motor seizures requires the anterior two-thirds of the corpus callosum. These studies (reviewed in Wada103) are of particular interest because, despite some important differences, the photosensitive baboon is believed to be a closer model of human epilepsy than are lower animals such as rodents and fowl.
Regulation of the seizure threshold and of seizure spread in different animal seizure types depends on seizure-gating pathways, which themselves are not pathways for seizure spread. Gating mechanisms include the substantia nigra and related circuits, ascending noradrenergic pathways, thalamic circuits, and some arising in the cerebellum (for a review see Gale37).
Intracellular events during GTCS have not been studied extensively, but neocortical neurons recorded during convulsive seizures induced by focal penicillin application show prolonged depolarization coinciding with the tonic phase, followed by sequential rhythmic depolarization and repolarization during the clonic phase. Gloor46 noted that GTCSs are associated with activation of N-methyl-D-aspartate (NMDA) receptors, which would permit calcium (Ca2+) influx into the neuron, and that many deleterious intracellular events follow the massive Ca2+ influx associated with convulsive seizures, some of which depend on activation of second messengers and some of which are direct. These can be transitory and manifested clinically by symptoms such as postictal stupor or Todd paralysis. However, irreversible cell damage or cell death may occur, especially with prolonged seizures or with status epilepticus (SE).57
Clinical Features
Ictal
Generalized convulsive seizures occur in several distinct epilepsy syndromes. Although the convulsion seems stereotyped, details of the seizure onset are important in the diagnosis of the related epilepsy syndrome. These may not be obtained from witnesses: video-EEG monitoring may be needed to document clinically relevant features, such as partial onset of a secondarily generalized event. Frontal lobe seizures particularly may imitate GTCSs (see the next section), and GTCSs may imitate focal seizures.36
Some patients reliably report a prodrome, premonitory symptoms hours or days before a seizure. It is surmised that these represent changes in cortical excitability or manifestations of some factor that also alters the seizure threshold. These are not auras; that is, they are not simple partial seizures that come immediately before more elaborate partial seizures, and they do not indicate a focal seizure onset. Common prodromes include mood changes, sleep disturbances, lightheadedness, anxiety, difficulty concentrating, and irritability.
GTCSs may begin with myoclonic jerks (Fig. 1) or, rarely, with absences.78,83 Before the tonic–clonic phase, versive movements of the head and eyes may occur. Although controversial and usually considered typical of partial seizures, versive movements clearly occur at or near the onset of some seizures which, as far as can be known, are generalized from the start.21,85,91 More complex circling behavior has also been documented.39,70 GTCSs can also occur at the onset, in the midst of, or at the end of absence status.6
The tonic phase40 begins with a brief flexor spasm of axial muscles that spreads quickly to the arms and legs, accompanied by loss of consciousness. The eyes deviate upward, the pupils dilate, and the jaw is rigid and partly open. This is followed by more prolonged tonic extension, spreading from axial muscles to the limbs. The initial closing of the mouth often results in tongue biting or other trauma, and the strong tonic contraction of respiratory muscles results in the characteristic cry. Apnea begins at the very onset of the tonic phase and can cause progressive cyanosis. Autonomic signs are prominent: Pulse and blood pressure rise, and profuse sweating and tracheobronchial hypersecretion are common. Although urinary bladder pressure rises, voiding does not occur because of sphincter muscle contraction.
The clonic phase begins gradually as a diffuse tremor (European authors often refer to this as vibratory), which slows from 8 to 4 Hz. It then emerges with cycles of inhibition interrupted by reappearance of the tonic contraction, producing atonia alternating with repeated violent flexor spasms. Each spasm is accompanied by pupillary contraction and dilation. The atonic periods gradually become longer until the last spasm. Voiding may occur at the end of the clonic phase as sphincter muscles relax. The convulsion typically lasts 1 to 2 minutes.
Postictal
Respiration returns almost at once after the last clonic jerk. Usually, the muscles are relaxed postictally, but a diffuse tonic state may return, resembling decerebrate rigidity and possibly causing further injury. The patient gradually awakens, often after a period of stupor or sleep, and is often confused, with some automatic behavior. Headache and muscle pain are common. The patient does not recall the seizure itself.
Complications of GTCS include trauma to the tongue, lips, and cheeks from trismus; head trauma due to a fall or to repeated banging of the head against hard surfaces during the clonic phase; vertebral compression fractures (usually asymptomatic); aspiration pneumonia; and, infrequently, neurogenic pulmonary edema, which may be related to the intense autonomic activity during the seizure. Sudden death in epilepsy, an uncommon but well-known event, may be related to an immediately preceding seizure. A GTCS can, rarely, be lethal74 (this topic is extensively reviewed in Lathers72). Langan et al.71 found that sudden death was more likely if GTCS had occurred in the previous 3 months (odds ratio 13.8, 95% CI: 6.6–29.1) (see Chapter 189). Pulmonary edema, apnea, cardiac arrhythmia, and aspiration with asphyxia have been proposed as mechanisms (see Chapter 10). Transitory metabolic changes after GTCS are usual. Changes in circulating hormone levels are also common.90 They are summarized in Tables 1 and 2. The elevation of plasma prolactin reaches its zenith of 5 to 30 times the upper limit of normal about 20 minutes after the seizure and, if present, almost certainly excludes pseudoseizure as the cause of a clinical episode. The postictal value should be compared to a control value obtained 24 hours later with no intervening seizure. Other hormone levels also rise after a seizure, but the relatively minor diurnal variation of prolactin levels and the relative ease of measuring them make this a practical test for clinical use. A prolonged convulsion may be associated with a minimal increase in the cerebrospinal fluid (CSF) cell count of 1 to 2 cells/mm3 and SE with a greater pleocytosis, but whatever the number of seizures, a cell count of more than 10 cells/mm3 should be taken as evidence of intracranial inflammation until proven otherwise.32
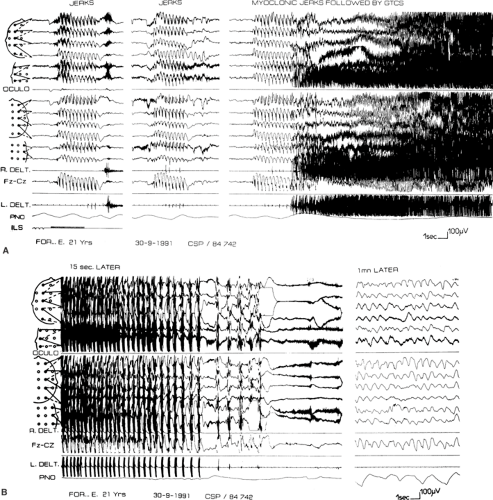 FIGURE 1. A generalized tonic–clonic seizure preceded by myoclonic jerks in a 21-year-old with juvenile myoclonic epilepsy. A: Three salvos of myoclonic jerks on awakening, the first elicited by intermittent photic stimulation. The others occur spontaneously, and the third is followed immediately by the tonic phase of the generalized convulsion. B: Clonic phase and end of the convulsion. Bilaterally synchronous jerks of decreasing frequency are recorded in the deltoid muscle EMG, followed by relative flattening of the EEG, partly masked by EMG artifact caused by postictal tonic spasm. Diffuse delta activity is recorded 1 minute later. The respiratory monitor records apnea during both the tonic and clonic phases of the seizure. R. delt, right deltoid; L. delt, left deltoid; PNO, pneumogram; ILS, intermittent light (photic) stimulation. |
EEG Manifestations and Clinical Correlations
Interictal
The waking EEG of patients with only GTCS is often normal.17 Slight nonspecific abnormalities of background activity may occur, and antiepileptic drugs (AEDs) may cause mild diffuse slow activity, especially at high levels. Barbiturates and benzodiazepines commonly cause diffuse increased β-wave activity. Some patients, especially those with a history of absences, may have some paroxysmal frontal intermittent rhythmic delta activity. Although abnormal in the EEG of the awake adult, this pattern reflects only a nonspecific disturbance of cerebral activity and is not considered epileptiform (reviewed in Zifkin109).
Generalized EEG interictal epileptiform abnormalities consist of spikes, sharp waves, polyspikes, and polyspike or spike-and-wave complexes (SWCs). Hyperventilation is often effective in bringing out generalized epileptiform activity, especially typical SWCs. Sleep recordings, ideally obtained without sedation, often increase the yield of epileptiform activity. Epileptiform activity may be reduced or abolished by chronic treatment with certain AEDs, especially valproate and the benzodiazepines. The type of interictal epileptiform activity can be related to the syndrome in which GTCS occurs:
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


