Chapter 71 Genetic and Metabolic Disorders of the White Matter
Introduction
Over the past two decades, an increasing number of novel heritable disorders affecting the white matter of the brain, or leukodystrophies, have been described, often with identification of a causative gene (Table 71-1). Pathognomonic magnetic resonance imaging (MRI) patterns (see Figure 71-1) or clinical characteristics permit identification in a number of these disorders, and should guide the clinician’s molecular diagnosis for many conditions (see Figures 71-3, 71-5, and 71-6 for MRI features). Challenges remain, however, for the neurologist, geneticist, or primary-care provider evaluating patients with suspected genetic or metabolic disorders of the white matter. In many settings, over half of subjects with suspected heritable white-matter disease do not receive a diagnosis, and therefore the focus of this chapter is on assisting the treating neurologist in the diagnosis of inherited disorders of the white matter.
Table 71-1 Molecular Causes of Leukodystrophies and Leukoencephalopathies (a Nonexhaustive List)
| Disorder | Gene |
|---|---|
| Acyl-coenzyme A (acyl-CoA) oxidase deficiency | ACOX |
| Adenylosuccinate lyase deficiency | ADSL |
| Aicardi–Goutières syndrome (AGS) | TREX1, SMHD1, RNAseH2A, B, C |
| Alexander’s disease (AxD)* | GFAP |
| Autosomal-dominant leukodystrophy with autonomic dysfunction* | Duplication LaminB1 |
| Canavan’s disease | ASPA |
| Cerebrotendinous xanthomatosis (CTX) | CYP27A1 |
| D-bifunctional protein deficiency | HSD17B4 |
| eIF2B-related disorder/VWM disease* | EIF2B1-5 |
| Glutaric aciduria type II/multiple acylCoA dehydrogenase deficiency (MADD) | ETFA, ETFB, ETFDH |
| 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase deficiency | HMGCR |
| Hypomyelination with congenital cataracts (HCC) * | FAM126A |
| Infantile sialic acid storage disease (ISSD) | SLC17A5 |
| Krabbe’s disease | GALC |
| Leukoencephalopathy with brainstem and spinal cord involvement and elevated lactate (LBSL)* | DARS2 |
| Lowe’s syndrome (oculocerebrorenal syndrome of Lowe, OCRL) | OCRL |
| Megalencephalic leukoencephalopathy with subcortical cysts (MLC)* | MLC1 |
| Metachromatic leukodystrophy (MLD) | ARSA |
| Mitochondrial neurogastrointestinal encephalopathy (MNGIE) | TYMP |
| Mucolipidosis IV | MCOLN1 |
| Oculodental digital dysplasia (ODDD)* | GJA1 |
| Pelizaeus–Merzbacher disease (PMD)* | PLP1 |
| Pelizaeus–Merzbacher-like disease (PMLD)* | GJC2 |
| Peroxisomal thiolase deficiency | ACAA |
| Polymerase gamma 1 (POLG1)* | POLG1 |
| Polyglucosan body disease (PGBD) | GBE1 |
| RNAse T2-deficient leukoencephalopathy* | RNASET2 |
| Sjögren–Larsson syndrome | ALDH3A2 |
| X-linked adrenoleukodystrophy (XALD) | ABCD1 |
| 18q minus syndrome | Deletion of short arm chromosome 18 |
VWM, vanishing white matter
* No specific biochemical marker clinically available, and molecular testing is primary diagnostic tool.
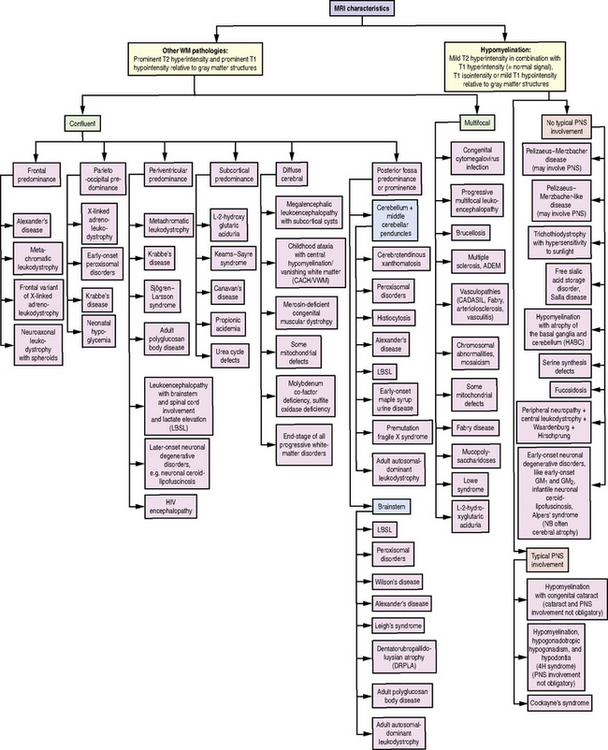
Fig. 71-1 Diagnostic algorithm for use in patients with abnormal myelination by MRI.
HIV, human immunodeficiency virus; PNS, peripheral nervous system; WM, white matter.
(Used with permission from Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology 2009;72:750–759.)
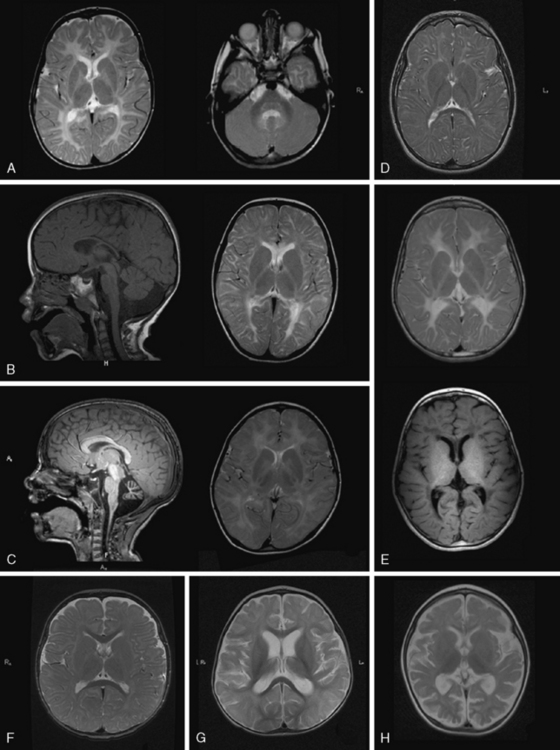
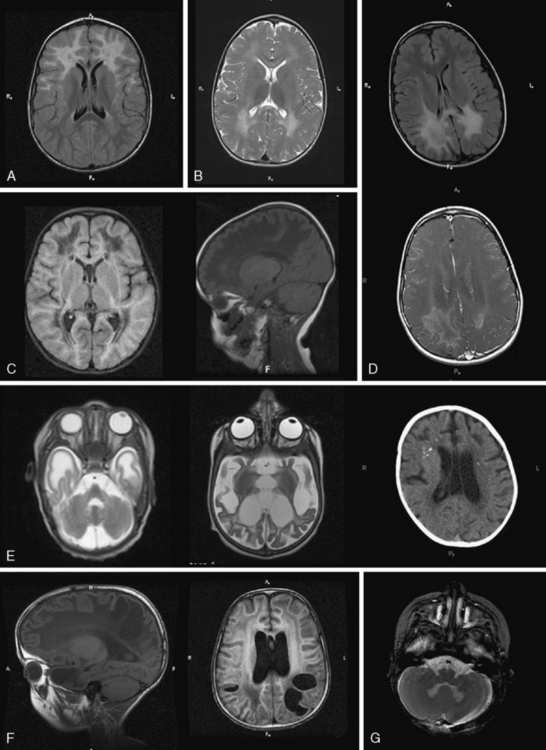
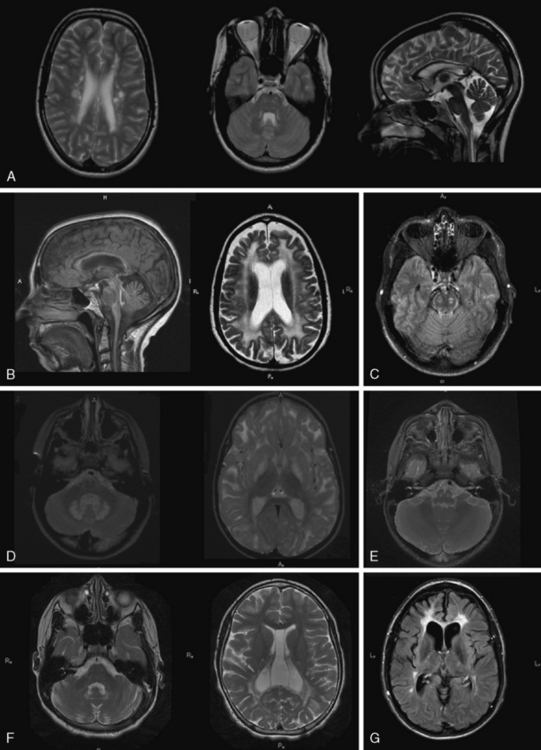
Fig. 71-6 Dysmyelinating and demyelinating leukodystrophies and pathognomonic MRI features, part II.
Given the heterogeneity and limited specificity in clinical findings, MRI pattern recognition is the most useful tool for evaluating suspected leukodystrophy patients [Schiffmann and van der Knaap, 2009] (Figure 71-1). MRIs should be reviewed comprehensively, with attention to changes over time, expected myelin development for the patient’s age, and characteristics of T1- and T2-weighted signal abnormalities. Broadly, there are two groups of leukodystrophies. Hypomyelinating leukodystrophies show increased white-matter signal on T2 relative to age, and often isointense or hyperintense white-matter signal on T1. Conversely, demyelinating leukodystrophies show increased white-matter signal on T2 relative to age, and hypointense white-matter signal on T1. Additional radiologic features should be examined, such as white-matter vacuolization or cysts, best seen on fluid-attenuated inversion recovery (FLAIR) or similar imaging paradigms; involvement of the basal ganglia, brainstem, cerebellum, or spinal cord; and abnormalities of cortical gray matter. Finally, findings consistent with calcifications should be noted, and if clinically indicated, a computed tomography (CT) scan or calcium-sensitive MRI sequences should be acquired to exclude the presence of calcifications that might be missed on standard MRI.
Hypomyelinating White-Matter Disorders
White-matter disorders with hypomyelination are relatively frequent; hypomyelinating leukodystrophies comprise 20 percent of leukodystrophies. They are characterized by a significant and permanent deficit of myelin [Schiffmann and van der Knaap, 2009]. Even with thorough genetic investigation, at least half of these hypomyelinating disorders lack definitive diagnosis, prognostic information, and potential for prenatal diagnosis. While most of these disorders show autosomal-recessive inheritance, both X-linked inheritance and de novo mutations are possible. Clinical manifestations common to hypomyelinating disorders are ataxia, spasticity, and nystagmus. Other non-neurological features can be valuable in diagnosis, such as hypodontia in 4H syndrome and cataracts in hypomyelination with congenital cataracts.
The diagnosis of hypomyelination may be made if two MRIs at least 6 months apart after the age of 12 months show little or no myelin development. Images in hypomyelinating leukodystrophies demonstrate diffusely hyperintense signal of the supratentorial white matter in T2-weighted images, and iso- or hyperintense white-matter signal on T1-weighted images. Certain areas, such as the posterior internal capsule, may have more normal-appearing myelin signal. Myelin deposition in infratentorial structures is usually higher. A definitive diagnosis of hypomyelination is not possible in young infants, as myelination is incomplete. If no myelin deposition is visible on the first MRI of a child older than 24 months, however, hypomyelination is highly probable [Schiffmann and van der Knaap, 2009].
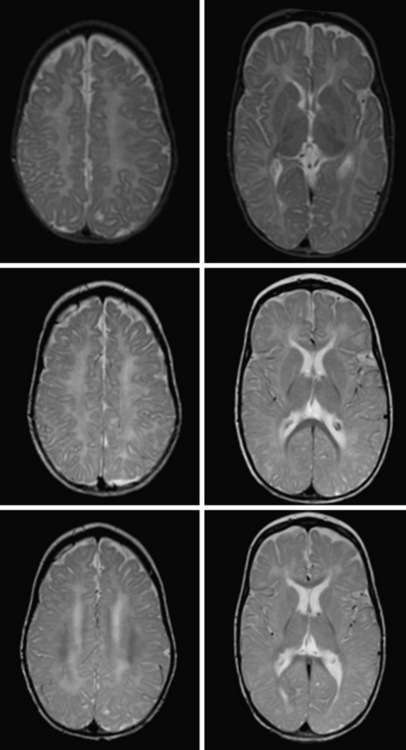
Delayed myelination is sometimes misdiagnosed as hypomyelination. In contrast to hypomyelination, in delayed myelination, myelination progresses on serial MRI images to near-normal myelin development (Figure 71-2). Gray-matter disorders with early onset often show hypomyelination, presumably a result of defective axonal function. Features such as early atrophy and signal changes of cortex and basal ganglia may help distinguish between primary hypomyelination and hypomyelination secondary to neuronal dysfunction (see Figure 71-3F, G, and H below).
In cases of primary hypomyelination, the disorders described below should be considered.
Pelizaeus–Merzbacher Disease
Pelizaeus–Merzbacher disease (PMD, OMIM 312080) is the prototypic hypomyelinating disorder, and is caused by alterations in the proteolipid 1 (PLP1) gene [Hudson et al., 1989; Trofatter et al., 1989]. Located on Xq22.2, this gene encodes the PLP1 protein, which constitutes roughly half of all myelin protein. The most common abnormality is a duplication of the entire gene, found in 60–70 percent of PMD cases and associated with the classic form of the disease [Ellis and Malcolm, 1994; Sistermans et al., 1998]. The lack of common breakpoints in these duplications results in varying sizes of the duplicated area between patients. Missense mutations account for 10–15 percent of cases. Several missense mutations affecting splicing or putative regulation of expression have also been described. Deletions are also seen in a smaller number of cases. Triplications are present in 1–2 percent of cases; higher copy numbers have been described in one child. More complex chromosomal rearrangements involving PLP1 or its promoter region have been described in individual cases [Muncke et al., 2004]. Deletions or null mutations are rare. Roughly 15 percent of cases fulfilling diagnostic criteria for PMD do not possess identifiable PLP1 mutations, suggesting the possibility of mutations in regulatory regions beyond genetic assays.
PMD is allelic with a relatively mild disorder, X-linked spastic paraplegia type 2 (SPG2), also caused by PLP1 mutations [Saugier-Veber et al., 1994]. In its pure form, the sole symptom associated with SPG2 is slowly increasing spasticity, more of the legs than of the arms, which begins during childhood or adolescence. In the complicated forms, additional symptoms, such as nystagmus, ataxia, dysarthria, and mild cognitive impairment, are present. Life expectancy in this form is normal. Peripheral neuropathy is another possible finding with certain PLP1 mutations. Altogether, alterations of PLP1 mutations give rise to a spectrum of disorders, ranging from the very severe connatal phenotype to mild spastic paraplegia with adolescent onset.
Genotype–phenotype correlations have been established for most PLP1 alterations, although clinical heterogeneity among patients with common genetic alterations makes definitive characterization difficult. Symptoms do not correlate with duplication size, but high copy number appears to predict increased clinical severity. Severe epilepsy has been reported in children with triplications, an otherwise uncommon feature of PMD [Wolf et al., 2005]. Point mutations are associated with the full spectrum of PLP1-connected disorders [Cailloux et al., 2000]. Demyelinating neuropathy is associated with either null mutations or mutations in PLP-specific regions. Patients with functional null mutations show relatively mild clinical course; they usually achieve independent, albeit clumsy, ambulation, and show mild cognitive impairment and demyelinating neuropathy. As spasticity increases after the first decade, patients become wheelchair-bound and develop pseudobulbar palsy. Histopathologic investigations show evidence of length-dependent axonal degeneration in these patients and in corresponding mouse models [Garbern et al., 2002].
PLP1 is a highly conserved, hydrophobic membrane protein with four transmembrane domains and a large cytosolic loop between the second and third transmembrane domains. The N- and C-termini are located in the cytoplasm. Different splicing of PLP1 yields a second, smaller isoform, DM20. Both isoforms are primarily expressed within the CNS. While the function of PLP1 and DM20 remains poorly understood, mutations that leave DM20 intact are associated with relatively mild phenotype. Mutations associated with severe phenotype likely cause protein misfolding, which activates compensatory oligodendrocytic responses that ultimately cause oligodendrocyte apoptosis. The pathogenicity of PLP1 duplications is less well understood, although it has been speculated that overexpressed PLP1 in endosomal and lysosomal compartments depletes myelin rafts of lipids necessary for the production of functional myelin compounds [Garbern, 2007].
MRI in PMD shows hypomyelination, indicated by diffusely elevated white-matter signal on T2 (Figure 71-3A). Myelination is arrested and fails to progress on repeated scans. Some patients show a more mottled, “tigroid” signal, perhaps due to small myelinated areas surrounding blood vessels. In some patients, myelin deposition is seen in the posterior limb of the internal capsule or the optic radiations. Myelination is usually present in the brainstem and sometimes in the cerebellar hemispheres. Often – but not always – the pyramidal tracts in the brainstem show high T2 signal. At a young age, atrophy is not considerable, especially in the cerebellum, although white-matter volume may be decreased. The corpus callosum is thin, reflecting lack of myelinated axons. As children grow older, generalized atrophy develops. Proton MR spectroscopy reveals low choline, due to reduced membrane turnover in the absence of myelinating oligodendrocytes, and normal to elevated N-acetylaspartate (NAA), due to more densely packed axons. Patients with null mutations or deletions show more advanced supratentorial myelination. In SPG2 patients, MRI abnormalities are variable, ranging from diffuse mild hypomyelination to mild periventricular signal changes. Carrier females sometimes display small areas of elevated white-matter signal, but MRI is usually normal.
Diagnosis of PMD is based on typical clinical presentation, radiologic evidence of hypomyelination, and detection of PLP1 alterations. Routine laboratory and metabolic tests, including cerebrospinal fluid (CSF), are normal. As most PLP1 alterations are duplications, simple sequencing must also be accompanied by gene dose quantification by Southern blot, quantitative polymerase chain reaction (PCR), or, more recently, multiple ligation-dependent probe amplification (MLPA). Recently, elevation of the dipeptide N-acetylaspartylglutamate (NAAG) has been described in several PMD cases, with NAAG level appearing to predict clinical severity [Burlina et al., 2006]. The mechanism of NAAG elevation, however, remains unknown, and it is neither specific to PMD, nor a marker of hypomyelination.
Pelizaeus–Merzbacher-Like Disease
PMLD is a genetically heterogeneous disease. In most cases, no gene has been identified. In a small subset of PMLD patients (fewer than 10 percent), mutations in GJC2 (also called GJA12), coding for connexin 46.6 (Cx47), have been found (OMIM 608804) [Henneke et al., 2008]. Located on chromosome 1q41–q42, this gene was identified using a homozygosity mapping approach in a large consanguineous family [Uhlenberg et al., 2004]. Mutations were subsequently found in other, unrelated, patients, confirming GJC2 as the responsible gene. It remains the only gene identified in PMLD; other approaches to identify PMLD genes, such as sequencing candidate genes for structural myelin proteins, have so far been unsuccessful.
As in PMD, patients typically present with nystagmus apparent after the first few weeks of life. Motor development is delayed and children display ataxia during the first few years. Pyramidal signs and frank spasticity often develop later. Compared with boys with classic PMD, motor and cognitive performance in PMLD is better, especially during the first years of life, and children are often able to walk, some without support. However, patients show a more precipitous decline, with progressive spasticity, prominent pseudobulbar signs, and facial weakness reminiscent of myopathic face. Patients become wheelchair-bound in their teens. Epilepsy starting in late school age has been reported in a substantial proportion of cases, and may be more severe in isolated cases, a very unusual feature for boys with PMD. Mild demyelinating peripheral neuropathy revealed by nerve conduction studies has been described in some patients, although it does not influence the overall clinical manifestation. A recently described family carrying a homozygote missense mutation in GJC2 showed a phenotype of almost pure spastic paraplegia and minor cerebellar signs [Orthmann-Murphy et al., 2008]. There are no histopathological data on PMLD.
MRI shows hypomyelination, and prominent signal abnormalities of the pyramidal tract of the brainstem have been described (Figure 71-3B). Cerebellar atrophy is mild or absent, at least in the early stages; supratentorial atrophy with considerable white-matter loss is prominent in older patients [Wolf et al., 2007]. Brainstem atrophy is often seen in later stages. Routine metabolic investigations are normal. In CSF, NAAG is elevated, as it is in PMD [Sartori et al., 2008].
4H Syndrome
This recently described leukoencephalopathy (OMIM 612440) is also characterized by hypomyelination. Its name is derived from its three main clinical findings: hypomyelination, hypodontia, and hypogonadotropic hypogonadism [Wolf et al., 2005, 2007; Timmons et al., 2006]. The disorder is rare and a genetic locus has not yet been identified. As affected siblings of both sexes have been reported, inheritance is presumed to be autosomal-recessive.
Besides these neurological symptoms, hypodontia is the most prominent diagnostic sign (Figure 71-4). Eruption of deciduous teeth is delayed, and its order disturbed. Normally, the lower median deciduous incisors erupt first, followed by the upper median incisors. In 4H syndrome, the deciduous molars erupt first, followed by the incisors, and finally by the upper median incisors. Despite this delayed and disorganized eruption, the deciduous teeth are usually complete. In the permanent dentition, however, some teeth are missing. The incisors have an abnormal shape and often also a yellowish color. About 10 percent of patients show natal teeth, an otherwise very rare finding occurring only in 1 in 1000–3000 newborns.
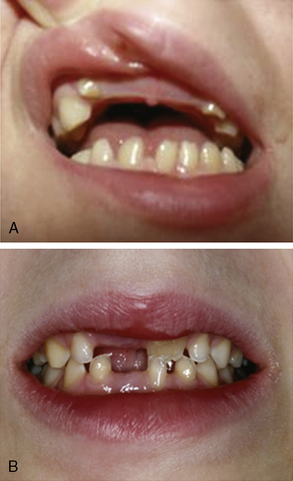
Fig. 71-4 Dental phenotype of children with 4H syndrome.
The upper median incisors erupt late. A, Child aged 5 years. The typical situation in a younger child with lacking upper median (and in this case also lateral) incisors. The left lower median incisor [Schiffmann et al., 1994] has not erupted yet. B, Child aged 8 years. No deciduous incisors have erupted. The right permanent upper median incisor [Paznekas et al., 2003] shows yellowish discoloration. The left median incisors have not erupted yet.
As the name of the disease suggests, MRI shows hypomyelinated white matter (see Figure 71-3C). T2 signal in the supratentorial white matter is diffusely elevated, with the exception of aspects of the posterior limb of the internal capsule and optic radiation. T1 white-matter signal varies from hypointense to hyperintense, depending on the amount of myelin deposited. Cerebellar white matter is usually myelinated on T2. Corpus callosum is thin. There is also early and considerable cerebellar atrophy, more of the vermis than of the hemispheres, in virtually all patients. Whether clinical severity correlates with amount of myelin deposited or degree of initial cerebellar atrophy has not yet been elucidated. Proton MR spectroscopy reveals low choline, as is usual in hypomyelination, and often also elevation of myo-inositol compatible with gliosis. In the later stages, considerable cortical atrophy and white-matter loss develop, indicating on-going myelin loss.
Metabolic investigations are all normal. In some children, a muscle biopsy, with assessment of respiratory chain enzymes, has been performed in the context of deterioration of symptoms with infections, but showed normal results. Analysis of the known genes involved in hypomyelination, including GJA1, in which mutations lead to oculodentodigital dysplasia, did not reveal abnormalities [Wolf et al., 2007].
Oculodentodigital Dysplasia
Oculodentodigital dysplasia (ODDD, OMIM 164200) is another hypomyelinating disorder characterized by dental abnormalities. Its inheritance is autosomal-dominant. Dominant mutations in another connexin gene on chromosome 6q21–23.2, GJA1, coding for connexin 43 (Cx43), cause ODDD. There is one family described with autosomal-recessive mutations leading to the same phenotype. Cx43 is expressed in the developing brain and teeth, and also in hands and feet [Paznekas et al., 2003].
Neurologic symptoms are common, although no large case series has been reported, so little is known about the exact time course and development of neurologic abnormalities. In childhood and adolescence, coordination problems and mild ataxia are common. In adulthood, slow neurologic deterioration is seen, with development of pyramidal tract lesions, ataxia, dysarthria, loss of bladder control, and finally, frank spasticity. Unsupported gait may become impossible in late stages. Optic atrophy and deafness are possible. Cognition is preserved in most patients, although learning disabilities have been reported. Whether and to what extent early cognitive deterioration occurs awaits further study. Peripheral nervous system appears unaffected [Loddenkemper et al., 2002].
Hypomyelination with Congenital Cataract
Affected patients present early with delayed motor development. Cognitive development is mildly to moderately delayed. Most HCC children learn to walk with support before their second birthday. Neurological examination reveals dysarthria, moderate to severe spasticity with elevated muscle tone, brisk reflexes, extensor plantar response, and cerebellar signs such as intention tremor and dysmetria. Nystagmus is rare. As in other hypomyelinating disorders, progression of secondary neurologic symptoms occurs, and children may be unable to walk, even with support, by the end of the first decade. An additional pathognomonic finding is congenital cataract [Zara et al., 2006]. Several HCC cases have been published, and one patient failed to develop cataract until age 9. A small subset of HCC patients suffers from occasional seizures. Peripheral neuropathy is seen in almost all patients, leading to loss of previously exaggerated tendon reflexes and distal muscle wasting. It is not yet known whether a broad phenotypic spectrum exists in HCC, and clinical presentation is heretofore remarkably homogeneous, with the exception of cataract and peripheral neuropathy, which are not present in all patients. There are no data yet about CNS pathology, nor experimental models to elucidate Hyccin function.
MRI shows hypomyelination, as evident from the diffusely elevated T2 white-matter signal. In contrast to those with other hypomyelinating disorders, HCC patients show additional areas of higher T2 white-matter signal and decreased signal intensity on corresponding T1-weighted images, indicating elevated water content in these areas, particularly in periventricular regions [Rossi et al., 2007] (Figure 71-3E). Cerebellar atrophy is not seen. In the early stages, normal myelin signal may be apparent in subcortical fibres and corpus callosum. In the late stages, white matter appears shrunken, with increased apparent diffusion quotients. Proton MR spectroscopy gives variable results, depending on the disease stage. Choline may even be slightly elevated in the early stages, which is unusual for a hypomyelinating disorder, and decreased in later stages.
Hypomyelination with Atrophy of the Basal Ganglia and Cerebellum
Hypomyelination with atrophy of the basal ganglia and cerebellum (HABC, OMIM 612438) is a rare disorder of unknown genetic origin [van der Knaap et al., 2002]. As no affected siblings of children with HABC have been reported, it is possible that this entity arises from a dominant de novo mutation, although autosomal-recessive inheritance remains possible. Disease severity ranges from severely affected infants presenting shortly after birth to more mildly affected children with initially normal development. In severe cases, children fail to achieve motor milestones and linguistic development, and show profound axial hypotonia. Some have nystagmus, and optic atrophy is possible. Spasticity is common. Extrapyramidal symptoms, comprising dystonia, rigidity, and choreoathetosis, are uniquely common to HABC relative to other white-matter disorders. Failure to thrive and microcephaly are common. In mildly affected children, unsupported walking is achieved within the first few years of life, sometimes on time, but is later lost. There is a combination of spasticity, extrapyramidal symptoms, and ataxia. cognitive impairment is mild, although patients tend to show cognitive decline in addition to deteriorating motor functions.
Recently published neuropathologic findings from one HABC patient detail slightly reduced white-matter volume, reduced oligodendrocytes, severe myelin deficiency, especially of deep white matter, and some white-matter loss, as indicated by the presence of macrophages in perivascular regions [van der Knaap et al., 2007]. Axons appeared relatively preserved. There was mild astrocytosis and a strong presence of microglia. The putamen was visible only as a small streak, and microscopy revealed substantial neuronal loss. Cerebral cortex was normal, both macroscopically and microscopically. In the atrophic cerebellum, there was loss of granule cells. Pyramidal tracts appeared degenerated in the brainstem and spinal cord. Metabolic investigations are normal in these children.
Sialic Acid Storage Disorders
Salla disease (OMIM 604369) and infantile sialic acid storage disease (ISSD, OMIM 269920) are both caused by autosomal-recessive mutations in SLC17A5, coding for Sialin, a lysosomal membrane protein transporting sialic acid from lysosomes [Verheijen et al., 1999]. The gene is located on chromosome 6q14/15. Recently, it was shown that Sialin also functions as a shuttle for aspartate and glutamate in synaptic vesicles [Miyaji et al., 2008]. Free sialic acid accumulates in lysosomes of many cell types, including liver and kidney cells and cultured fibroblasts. In leukocytes, this accumulation causes vacuoles visible at light microscopic examination. Electron microscopy reveals membrane-bound vacuoles filled with fibrillogranular amorphous material. The pathogenesis of free sialic acid storage disease and the role of sialic acid remain unclear.
Both diseases are characterized by elevated excretion of free sialic acid in urine. Sialic acid is also elevated in other fluids, such as CSF. Recently, two siblings have been described who lack the characteristic sialuria; sialic acid was elevated only in CSF [Mochel et al., 2009]. Prevalent in Finland, Salla disease is characterized by seemingly normal early development, followed by presentation with hypotonia and ataxia in the second half of the first year of life. Nystagmus is also common. It may be evident in the neonatal period and frequently disappears. Many children also show strabismus. Spasticity develops slowly, and mild extrapyramidal symptoms are common in later stages. Mean age at walking is 4 years, with roughly one-third of patients who do not develop independent ambulation. Language is severely affected and usually dysarthric; patients are, at best, able to produce short sentences. Epilepsy with short, complex focal seizures is relatively common. In some patients, there is evidence of peripheral hypomyelination with decreased nerve conduction velocities. The disease is stable over a long period of time, with ultimate late progression. Additional symptoms may include short stature or hypogonadotropic hypogonadism. Facial features become coarse in adulthood. Otherwise, there is no evidence for dysostosis multiplex or hepatosplenomegaly, despite the presence of free sialic acid storage in liver and spleen. Life expectancy is normal [Aula et al., 2000].
A phenotype with intermediate severity relative to Salla disease and ISSD has also been identified.
MRI in patients with Salla disease shows hypomyelination [Sonninen et al., 1999] (Figure 71-3D). There is white-matter volume loss. Corpus callosum may be stringlike, especially in severe cases. There is usually cerebellar atrophy, and supratentorial atrophy is found in older patients. Thickening of the calvarium is another common radiologic feature. Proton MR spectroscopy reveals a high NAA peak, likely due to elevated free sialic acid, whose N-acetyl peak co-resonates with the N-acetyl peak of NAA [Varho et al., 1999].
Fucosidosis
MRI shows hypomyelination. A characteristic feature of fucosidosis is high T1 and low T2 signal in globus pallidus, thalamus, and substantia nigra. Cerebral and cerebellar atrophy may be prominent in older patients [Prietsch et al., 2008].
Serine Synthesis Defects
Children with 3-phosphoglycerate dehydrogenase deficiency are born microcephalic, and their development is grossly delayed. Epilepsy develops in the second half of the first year of life; West’s syndrome is one possible manifestation. Supplementation with serine and glycine is effective in seizure management. If treatment is commenced prenatally, head circumference at birth and development are normal [de Koning et al., 2004]. In untreated children, MRI shows hypomyelination and white-matter volume loss [de Koning et al., 2000]. Corpus callosum is thin and short. Myelination improves under treatment.
The first documented patient with phosphoserine aminotransferase deficiency presented in the neonatal period with severe epilepsy resistant to medical treatment and rapidly developing microcephaly. MRI showed supratentorial and brainstem atrophy. Supplementation with serine and glycine did not attenuate the seizures. The same treatment, if started before the development of symptoms, was shown to prevent epilepsy and enable normal development in the sibling of the proband [Hart et al., 2007].
Cockayne’s Syndrome and Trichothiodystrophy
Cockayne’s syndrome (CS) is a rare disease combining neurologic and non-neurologic features. This disorder and related disorders of DNA repair, such as cerebro-oculo-facial syndrome (COFS) and trichothiodystrophy (TTD), are genetically heterogeneous and are caused by mutations in CSA (CKN1 or DNA excision repair protein ERCC-8, responsible for 20 percent of CS cases), CSB (CKN2 or ERCC-6, responsible for most of the remainder of CS cases), XPB (ERCC3, OMIM 610651), XPD (ERCC2), XPG (ERCC5), ERCC1-XPF, TTDA and TTDN1 genes, and possibly others [Weidenheim et al., 2009].
The classic form, Cockayne’s syndrome type I, presents in the first year of life with failure to thrive; weight is more affected than length (“cachectic dwarfism”), and there is loss of subcutaneous fat, leading to a “wizened,” bird-like, progeroid face. Microcephaly also develops, usually by the end of the second year. Children develop contractures of the large joints, giving them a typical posture. Hands and feet are disproportionally large. Dental caries is prominent. Psychomotor development is also delayed, resulting in mild to severe cognitive impairment. Predominant neurologic features include ataxia and spasticity, which show slow progression. In late stages, peripheral neuropathy leads to muscle wasting and loss of the initially increased tendon reflexes. Over half of individuals develop sensorineural hearing loss. Most suffer from pigmentary retinal degeneration and cataracts. Autonomic dysfunction (hypolacrimia, hypohydrosis, miosis, acrocyanosis) is possible. In Cockayne’s syndrome type II, the clinical picture is much more severe, with growth failure already evident at birth. Loss or even absence of subcutaneous fat is striking. Joint contractures and kyphosis develop rapidly. Hypotonia is prominent initially, followed by development of spastic tetraparesis. Psychomotor development is absent or extremely delayed, with subsequent deterioration and early death. Subcutaneous fat loss has been treated in both types by early hypercaloric tube feeding, which allows reasonable growth in some children. Type III describes patients with milder forms of disease. Cutaneous photosensitivity is also characteristic of CS, occurring in 75 percent of all patients [Rapin et al., 2000].
Both syndromes are caused by defective nucleotide excision repair. Over 30 proteins are involved in this process. It eliminates DNA lesions induced by ultraviolet light. There are two major subpathways of nuclear excision repair: transcription-coupled repair, dealing with reparation of transcribed genes, and global genome repair, removing lesions in the entire genome. Defective DNA repair can be demonstrated by irradiating cultured skin fibroblasts with ultraviolet light and subsequently measuring unscheduled DNA synthesis. This unscheduled DNA synthesis is diminished in TTD and xeroderma pigmentosum patients. In CS, this unscheduled DNA synthesis is not significantly attenuated, but the otherwise rapid recovery of RNA and DNA synthesis after ultraviolet irradiation is adversely affected, indicating that the global genome repair is still functional. Additionally, reduction in basal transcription is also seen, an important signal for apoptosis. These defects of transcription, perhaps combined with activation of apoptosis, are thought to be mainly responsible for the neurologic symptoms in CS. Complementation assays in cultured cells could distinguish two different complementation groups, CS type I (OMIM 216400), caused by mutations in the gene coding for group 8 excision-repair cross-complementing protein (ERCC8), and type II (OMIM 133540), due to mutations in ERCC6. TTD is also heterogeneous, genetic defects having been identified in at least three different genes [Cleaver et al., 2009].
MRI of patients with CS shows hypomyelination, its degree corresponding to clinical severity. In severe cases with CS type II, hypoplasia of cerebellum and brainstem is possible. Basal ganglia calcifications are common. Similar features are seen in TTD, although calcifications are less common than in CS [Rapin et al., 2000; Adachi et al., 2006].
18q Minus Syndrome
In this disorder (OMIM 601808), the distal region of the long arm of chromosome 18 is deleted. It occurs de novo most commonly. The contiguous gene deletion usually involves the bands 18q22.3→qter. The gene for myelin basic protein (MBP), a component of healthy myelin, is located within this region. It has been postulated that haploinsufficiency for MBP leads to the myelin abnormalities observed in 18q minus syndrome. Heterogeneity in severity of clinical symptoms and hypomyelination between patients, despite consistent loss of MBP, is a focus of on-going inquiry. There is a well-investigated mouse model, the shiverer mouse, with homozygous rearrangements in the MBP gene [Nave, 1994], which lead to CNS, but no peripheral, hypomyelination. It is unknown why peripheral myelin is spared, despite MBP expression in peripheral nerves.
MRI shows hypomyelination of variable, but usually mild, degree. The myelin signal in the cerebral hemispheres may be inhomogeneous, with patchy white matter abnormalities. Corpus callosum may be thin. There may be mild supratentorial atrophy [Linnankivi et al., 2006].
SOX10-Associated Disorders
These rare syndromes are caused by mutations in SOX10 on chromosome 22q13, which encodes a transcription factor for various genes, some involved in myelin formation and metabolism, such as GJB1 (connexin 32). These disorders are characterized by a white hair lock and hypomelanotic spots, sensorineural deafness, and Hirschsprung’s disease. Patients are affected with varying severity, ranging from antenatal onset with congenital arthrogryposis multiplex and severe neurologic abnormalities, to more mildly affected patients who lack neurologic manifestations (Waardenburg–Shah syndrome, WS4, OMIM 277580). The severe variant has been designated peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg’s syndrome, Hirschsprung’s disease, or PCWH (OMIM 609136). Consistent with a neurocristopathy, many features of this disease can be explained by defective differentiation or migration of neural crest cells. In the severe cases, MRI reveals hypomyelination [Touraine et al., 2000]. The milder phenotype of WS4 is explained by SOX10 haploinsufficiency, whereas the severe form, PCWH, is thought to be caused by a dominant negative mechanism. Mutations found in children with PCWH are all truncating and located in the last exon. These mutations, unlike SOX10 mutations leading to WS4, escape nonsense-mediated decay and can therefore exert their dominant negative effect on protein level [Inoue et al., 2004, 2007].
Neurologic symptoms of children with PCWH include delayed development, nystagmus, spasticity, and ataxia. In severe cases, neonates are already symptomatic, with profound hypotonia, seizures, or congenital arthrogryposis due to hypomyelinating neuropathy. Other possible symptoms are reduced tear and saliva production, anhidrosis, and severe failure to thrive, in addition to the classic syndromes of WS4. Neuropathologic investigations of a severely affected infant have shown absence of central and peripheral nervous system myelin at the age of 3 months [Inoue et al., 2002]. Detailed MRI features have not been reported for many patients, but preliminary cases suggest mild to severe hypomyelination, and possibly atrophic brainstem [Inoue et al., 1999].
White-Matter Disorders with Demyelination
If MRI is not consistent with hypomyelination, there is white-matter hypointensity on T1 instead of iso- or hyperintensity, and there is hyperintensity on T2, the imaging pattern fits the demyelinating leukodystrophies. They comprise the leukodystrophies with primary demyelination, leukodystrophies with white-matter vacuolization, calcifying leukoencephalopathies, cystic leukoencephalopathies, leukoencephalopathies with brainstem involvement, and most adult-onset leukoencephalopathies. In the assessment of patients with these leukodystrophies, careful attention should be paid to specific neuroimaging features, including basal ganglia or brainstem signal abnormalities, contrast enhancement, cysts, calcifications, contrast enhancement, or specific FLAIR imaging abnormalities (Figure 71-5 and Figure 71-6; see also Figure 71-3) to assist in the differential diagnosis.
Primary Demyelinating Leukodystrophies
Alexander’s Disease
Alexander’s disease (AxD, OMIM 203450)[Alexander, 1949] is associated with mutations in the gene encoding the glial fibrillary acidic protein (GFAP) [Brenner et al., 2001]. GFAP mutations are thought to confer gain-of-function mutations, and a mutation on a single allele is sufficient to cause disease. In most cases, mutations are sporadic, although familial cases are described in adult- or juvenile-onset cases. In familial cases, inheritance is autosomal-dominant. There is usually concordance in presentation within a family, such that a family with adult-onset presentation and subsequent infantile presentation has not been reported. There is no definite genotype–phenotype correlation, with the exception of the two most common mutations, R79 and R239. R239, in particular, is associated with earlier onset and poor prognosis.
GFAP mutations are thought to result in decreased solubility of the glial fibrillary acidic protein, and accumulation of GFAP, along with vimentin, αβ-crystallin, and heat shock protein 27, result in Rosenthal fiber (RF) aggregation. RFs, accumulating in astrocytes, are thought to obstruct normal glial cell function and result in myelin destruction. RF accumulation in the ventricular collecting system is believed to cause the hydrocephalus seen in the clinical setting. Studies to improve pathophysiologic understanding of this disease and possible treatment strategies are on-going in an existing murine model of AxD. AxD is typically suspected from clinical presentation and characteristic MRI features. Typical AxD imaging features include frontal predominance of white-matter abnormalities, basal ganglia and or brainstem involvement, a periventricular rim of altered signal on T1- and T2-weighted imaging, and contrast enhancement of specific intracranial structures (see Figure 71-5A). The presence of four of five of these features makes diagnosis of AxD very likely and should prompt mutation testing [van der Knaap et al., 2001]. In very young infants and in the juvenile- or adult-onset cases, MRI may be less typical and involve only restricted brain regions. Additional features described in AxD imaging include predominant or isolated involvement of posterior fossa structures, multifocal tumor-like brainstem lesions, brainstem atrophy, and garland-like abnormalities along the ventricular wall [van der Knaap et al., 2005, 2006]. MR spectroscopy can be helpful in the diagnosis if it shows a lactate peak in affected tissues, but may also lead to inappropriate evaluations for mitochondrial cytopathies. There have been reports of findings of elevations of GFAP protein in CSF [Kyllerman et al., 2005], but this test is not used as a clinical tool.
X-Linked Adrenoleukodystrophy
X-linked adrenoleukodystrophy (XALD, OMIM 300100) is associated with mutations in the ABCD1 gene [Mosser et al., 1993] encoding a peroxisomal membrane transporter. This disorder follows X-linked inheritance, and often, when male children are diagnosed with the childhood-onset cerebral form, disease manifestations are recognized in obligate female carriers or male relatives of the proband. Differences in disease manifestations are known to occur with identical genotype, and even within a sibship, underscoring the likely effect of other genetic factors on clinical presentation. Genotype does not predict very long chain fatty acid levels or specific clinical prognosis. Genetic changes reported include missense mutations, nonsense mutations, frameshift mutations, small deletions/insertions, and large deletions.
Of note, an allelic disorder, with a neonatal presentation of cholestasis, hypotonia, and developmental delay, is caused by a contiguous gene deletion syndrome involving the 5′ end of ABCD1. This disorder is called contiguous ABCD1 DXS1357E deletion syndrome (CADDS), and is clinically distinct from XALD [Corzo et al., 2002].
The estimated prevalence of XALD is estimated to be 1:20,000–1:50,000. The estimated prevalence of hemizygotes (affected males) and heterozygotes (carrier females) is estimated at 1:16,800 [Bezman et al., 2001].
The pathophysiology of XALD is believed to arise from accumulation of saturated very long chain fatty acids (SVLCFA) within the brain. This accumulation is thought to result from defective peroxisomal fatty acid oxidation of SVLCFA, caused by defective transport by the mutated ABCD1, possibly due to altered adenosine triphosphate (ATP) binding [Gartner et al., 2002; Roerig et al., 2001].
Diagnosis is based on characteristic clinical presentation and suggestive MRI features. MRI shows a predominance of occipital findings (see Figure 71-5D), although frontal and corpus callosum variants are recognized. The affected white matter appears hyperintense on T2 and hypointense on T1. Characteristically, there is a rim of enhancement around the abnormal tissue that can be very helpful in establishing the diagnosis, as few other leukodystrophies, with the exception of Alexander’s disease, show significant contrast enhancement. When XALD is suspected, appropriate clinical tests include fasting VLCFA testing on plasma, which shows an excess in SVLCFA with specific abnormalities in C26:0, C24:0/C22:0, and C26:0/C22:0 ratios [Moser et al., 1981]. Mutation testing of the ABCD1 gene provides molecular confirmation of the diagnosis, with attention to the 7 percent of cases with deletions or rearrangements.
Treatment with Lorenzo’s oil or diets rich in oleic and erucic acids, in addition to VLCFA restriction, can alter VLCFA levels. As treatment strategies for XALD evolve, interest in newborn screening is growing. This can be done using liquid chromatography-tandem mass spectrometry (LC-MS/MS) to test for the analyte 1-hexacosanoyl-2-lyso-sn-3-glycero-phosphorylcholine (26:0-lyso-PC)[Hubbard et al., 2009; Raymond et al., 2007]. As with many disorders now being considered for widespread newborn screening, the clinical benefit of early diagnosis remains to be established.
Treatment of XALD depends on the stage and type of disease manifestations, and is still research-based. Treatment with Lorenzo’s oil, which decreases hexacosanoic acid (C26:0), does not appear to stop or reverse cerebral disease once it has begun, although there are reports of improved long-term stability in presymptomatic patients in open-label studies with no placebo control [Moser et al., 2007]. The use of Lorenzo’s oil is investigational and should be performed in the context of a clinical research protocol. Bone marrow transplantation (BMT) is indicated in children with early-stage, cerebral-form XALD, as evidenced by active disease on MRI. Family members of an affected proband should be tested by VLCFA and monitored for early MRI evidence of cerebral involvement to identify candidates for BMT. These patients should be evaluated by clinicians with specific expertise in this disorder. Gene therapy is a potential future tool and research studies are under way. Supportive care can improve comfort and quality of life for XALD patients. Careful monitoring and treatment of adrenal insufficiency should be part of therapeutic management.
Metachromatic Leukodystrophy
Metachromatic leukodystrophy (MLD, OMIM 250100) [Greenfield, 1933; Von Hirsch and Peiffer, 1955; Suzuki and Chen, 1966] is caused by mutations in the ARSA gene encoding arylsulfatase A [Stein et al., 1989; Austin et al., 1964] on chromosome 22q13.31, and is inherited in an autosomal-recessive manner. Homozygous or compound heterozygous ARSA mutations impair arylsulfatase A degradation of sulfatides, causing sulfatide accumulation within the brain and peripheral nervous system. Complete loss of arylsulfatase A activity (“I” or “O” alleles) is typically associated with early infantile MLD. Partial loss of arylsulfatase A activity (“R” or “A” alleles) is typically associated with juvenile- or adult-onset MLD. Compound heterozygosity with an I and A allele usually results in juvenile-onset MLD. Rarely, subjects with microdeletions of 22q13 and deletion of the ARSA gene with an MLD-causing mutation on the other allele have been diagnosed with MLD.
MLD diagnosis is often suspected based on clinical manifestations of motor impairment with a peripheral neuropathy. Typical MRI features include sparing of arcuate fibers and a rim of subcortical white matter, with involvement of periventricular and deep white matter in the supratentorial CNS (see Figure 71-5B). Involved white matter takes on the appearance of radiating stripes that can be highly suggestive of the disorder and reflects accumulation of sulfatides in perivascular macrophages. These radiating stripes are also present in other disorders, however, including Krabbe’s disease. In early stages or in adult cases, incomplete neurologic findings can complicate diagnosis, and early involvement of the corpus callosum may provide a clue. Occasionally, isolated involvement of cranial or peripheral nerves has been seen in the early stages of disease.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


