Introduction
The success of molecular genetic strategies in finding genes underlying human monogenic epilepsies has set the stage for a new generation of defined experimental genetic models to unravel the mechanisms of inherited seizure disorders. While each discovery of a causative human epilepsy gene offers the promise of an accurate molecular diagnosis, identification of the mutant gene alone is unlikely to lead to advances in medical therapy until its cellular mechanism of action is at least partially understood. This is based on two simple reasons: First, mutations can alter gene function in various ways, most notably as a loss or a gain of biologic function, each requiring entirely different strategies for pharmacologic intervention. Second, the inherited gene defect is likely to trigger epilepsy through a complex sequence of downstream cellular changes in the developing brain, leading to the emergence of a specific seizure phenotype at a particular age. This developmental plasticity means that the destabilizing action of the gene on network excitability must be isolated from many other potentially misleading structural and functional alterations.
Genetic animal models offer the opportunity to experimentally dissect these manifold effects of the inherited disease gene in the developing brain, and to precisely define which alterations in brain circuit microanatomy and physiology directly contribute to the onset of seizures. Once this therapeutic target is obtained, a rational search for pharmacologic compounds to prevent or reverse the imbalance can begin. Since many different biologic pathways have now been implicated by known epilepsy gene mutations, it is hoped that similarities among the models may point to a smaller number of common synaptic pathways and membrane excitability defects that are shared among seizure syndromes.66 This principle of convergent epileptogenic defects for specific seizure types predicts that information from individually rare inherited epilepsies will one day be translatable into broad-spectrum antiepileptic drugs. In this sense, along with continuing gene discovery, the current efforts at target discovery (i.e., to precisely define the pathogenesis of epilepsy in genetic models at the molecular and cellular levels, rather than simply at the gene and electroencephalographic levels) will provide the long sought framework for major advances in the development of effective pharmacologic therapy for many inherited seizure disorders.
General Description
Two general categories of genetic models have been historically important. Over the past 50 years, the discovery of a lowered threshold to convulsant stimulation or actual spontaneous seizures in inbred strains of various species provided the substrate to search for brain mechanisms underlying epilepsy and to screen for anticonvulsant efficacy. Some of these original strains, which reflect the deleterious effects of many unidentified genes, have been preserved in colonies; others have been created de novo using recombinant inbred strain methods,29 a strategy that allows the genetic propagation of randomly combined sets of hyperexcitability alleles already present but asymptomatic in the “wild type” genetic background. These models all possess the virtue of displaying either spontaneous electrographic and behavioral seizures or a lowered threshold to seizures evoked by various modalities, and thus share polygenic susceptibility increases resembling those that contribute to many human seizure disorders. These inbred strains provide highly reproducible laboratory models for elucidation of anatomic seizure pathways and antiepileptic intervention.
A second, more powerful approach has been to survey animals for epileptic phenotypes linked to single locus mutations, either those which have occurred spontaneously, or increasingly, those engineered by recombinant DNA mutagenesis techniques.12,64,67 The latter are typically created by the insertion of additional copies of a gene to increase the expression levels of a gene product, the deletion of a specific gene by homologous recombination to create a loss of function of the targeted gene, or the alteration of a gene sequence to produce an entirely novel gain of function. A refinement of this approach is to replace a wild-type allele with one engineered by site-directed mutagenesis to achieve a desired effect upon the function of the protein, typically one that has been designed to exactly replicate a human mutation. Recreating the precise molecular lesion of a form of human epilepsy in another species, an orthologous gene model, provides an exceptional opportunity to understand the natural history of the clinical disorder.
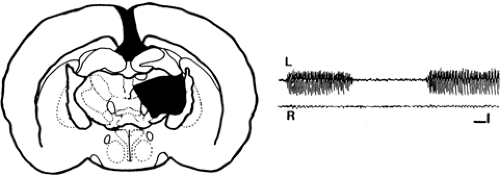 FIGURE 1. Ibotenic acid lesions of the thalamic reticular nucleus (nRT) abolish spike-wave discharges (SWDs) in the absence epilepsy model genetic absence epilepsy rat of Strasbourg. Left: Reconstruction of lesions induced by ibotenic acid, an excitotoxin that selectively kills neurons without affecting fibers of passage. Right: Electroencephalographic (EEG) recordings 4 days after unilateral nRT lesion show abolition of SWDs in the ipsilateral hemisphere, leaving only abortive low-frequency discharges. The animal was previously callosomized to prevent contralateral diffusion of the SWDs originating from the intact side. L, left EEG; R, right EEG. (From Avanzini G, Vergnes M, Spreafico R, et al. Calcium-dependent regulation of genetically determined spike and waves by the RTN of rats. Epilepsia. 1993;34:1–7.) |
While many inherited human epilepsies may ultimately prove to be multigenic in origin, defects at a single gene locus suffice to produce a stereotyped seizure disorder and can be isolated and studied on many different experimental genetic backgrounds. Since the earliest electrophysiologic studies of behavioral mutants in fruitflies and mice,36,39,68,103 spontaneous single gene mutations have contributed key examples of neuronal hyperexcitability mechanisms. Drosophila melanogaster hyperexcitability models of mutant ion channels, pumps, RNA binding proteins, and even KCC2-like cotransporters responsible for depolarizing GABA receptor responses continue to be described,31,34,72 and this system shows great promise for rapidly identifying modifier gene loci,33,88 an emerging frontier in epilepsy genetics. High-throughput mutagenesis protocols are now available to accelerate identification of relevant genes in other simple genomes, including zebrafish,5 round worms,101 and mice.32 Of all of these genetic systems, defined mutations in the mouse offer the most comprehensive opportunity to identify genes linked to spontaneous, electroencephalographically proven epilepsy, and provide a powerful system to investigate the pathogenesis of single locus mutant phenotypes discovered in human mendelian pedigrees. Regardless of the species of origin, novel gene candidates for hyperexcitability discovered
through these experimental models narrow the search for genes in a linkage region mapped for human positional cloning studies, and thereby greatly facilitate human epilepsy gene discovery.
through these experimental models narrow the search for genes in a linkage region mapped for human positional cloning studies, and thereby greatly facilitate human epilepsy gene discovery.
Questions That Can Be Addressed in Genetic Models of Epilepsy
Genetic animal models of epilepsy are useful to (a) define the nature, timing, and distribution of excitability changes leading to seizures in the developing brain; (b) implicate or exclude specific molecules, synapses, and networks in epileptogenesis as well as isolate gene modifiers that enhance or suppress seizures; (c) analyze the rate of progression and extent of seizure-induced neural damage during chronic epilepsy; and (d) evaluate the specificity of new antiepileptic therapies. While exact, mutation-specific (“knockin”) orthologous models of most known inherited human epilepsy syndromes are still under development, mouse models representing at least half of the known human epilepsy genes are currently available as targeted deletion (“knockout”) or overexpression mutants. These “first generation” models can mimic the overall effect of the mutation, namely, a loss or gain of function of the gene, but may not in fact reproduce the precise excitability phenotype seen in a given missense mutation of the same gene in a human proband. At the molecular level this is readily understandable, since a protein such as an ion channel that functions poorly yet continues to physically interact as a membrane protein with other channel subunits or intracellular signals often creates a different cellular defect than no protein at all. Nevertheless, these mutants provide general information on basic neural processes likely to be involved in the human clinical counterpart.
Brief Description of the Individual Models
A variety of inbred models that remain genetically uncharacterized have been of significant historical importance in the experimental analysis of epilepsy, including photosensitive baboons,62,91 and chickens92; however, with a few important exceptions, including a novel unstable dodecamer expansion repeat mutation of Epm2b (Nhlrc1) identified in dogs with canine Lafora disease,53 newer models are based on defined gene lesions in rodents. Due to the large number, this chapter will focus on recent work in a smaller number of exemplary models currently under study.
Table 1 Single Gene Models Displaying Absence Epilepsy in Mice | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
Generalized Absence Seizure Models in Inbred Strains
Two inbred rat models of absence epilepsy, the genetic absence epilepsy rat of Strasbourg (GAERS) and the Wag-Rij rat strain, continue to serve a seminal role in defining the networks and cellular excitability mechanisms of absence seizures emerging from aberrant thalamocortical oscillations.22,26 Inheritance of spike-wave (SW) epilepsy in these models is not due to a single gene locus, although potentially contributory gene variants can be identified that are absent in other unaffected rat strains, for example, a variant in the KCNK9 gene coding for the TASK3 (Twik-like acid-sensitive K+) channel in the GAERS model.35 Both inbred rat models exhibit spontaneously occurring bilaterally synchronous 5- to 11-Hz SW with behavioral arrest with a late onset at 3 to 4 months of age lasting into adulthood. The neocortex, thalamic relay neurons, and reticular thalamic nucleus are involved in the aberrant synchronous discharge, cognitive performance during the seizure is impaired, and the oscillations share major features of the circuitry and pharmacology, if not developmental onset, of slower 3/sec absence seizures in humans. Occasionally, the same discharge pattern is reported in other inbred rat strains.84
A number of important electrophysiologic and pharmacologic insights have been derived from these two strain models. Key among these are the involvement of the nucleus reticularis thalami in the expression of spike-wave epilepsy (Fig. 1) and the role of elevated low-voltage T-type calcium currents in this discharge.97 Additional control over thalamic oscillations is regulated by enhanced HCN1 gene expression and decreased cAMP responsiveness of a second pacemaking current, Ih, favoring rebound excitation in these neurons.11 Other recent findings point to regional blood flow increases,63 cortical excitability changes in deep layer neurons,23 a low threshold trigger zone in barrel cortex,58 and γ-aminobutyric acid (GABA)-ergic signaling impairments.6 These models have served as a mainstay for the analysis of thalamocortical spike-wave seizure modulation by a variety of receptor families, as well as a wide range of antiepileptic pharmacology.26
Generalized Convulsive Seizure Models in Inbred Strains
The genetically epilepsy-prone rat (GEPR) is a model of experimental audiogenic seizures, a form of reflex epilepsy that has been useful in tracing the brainstem pathways mediating high decibel-induced convulsions with wild running.41 While a rare clinical counterpart of this behavioral seizure type, epilepsia cursiva, has been recognized in humans,37 the model set the stage for analysis of audiogenic mouse mutant models with defined single gene lesions, including the serotonin 5HT2C receptor,8 the FMR1 Fragile X mouse,105 and the JAMS1 mouse locus59 that corresponds with a human locus for febrile seizures. Interestingly, a loss of function mutation in MASS1, another gene shown to underlie audiogenic seizure susceptibility,86 was subsequently found within a human pedigree with febrile seizures.61
The EL mouse inbred strain is notable for the development of seizures following repeated episodes of vestibular stimulation. Susceptibility begins at 10 weeks of age, and the seizures are manifest as running fits followed by convulsions of possible cortical origin.60,90 A variety of defects have been described in this model, and it continues to find a use in imaging GABAergic receptor signaling plasticity in chronic epilepsy.30 Another model of convulsions evoked by stress and handling has been described in various strains of the domestic Mongolian gerbil. This model was once used extensively in neuropharmacologic testing, and more recently has been found to exhibit changes within the GABA interneuron population.10
The spontaneously epileptic rat (SER) was developed by crossing the tremor and zitter strains and exhibits spontaneous tonic convulsions, absencelike seizures, and spongiform encephalopathy ending in lethality by 20 weeks of age. Recent studies add abnormalities in glutamate transport to a diverse set of other neuropathologic changes resulting from coexpression of these two loci.2 The zitter locus contains a splicing mutation in the gene encoding attractin, an enzyme with a critical role in myelination.46 As a model displaying two distinct seizure types, it finds use in studies of antiepileptic drug profiles.40
An inbred rat strain with a cortical band heterotopia and convulsions resembling the human double cortex malformation syndrome, the telencephalic internal structural heterotopia (Tish) rat, has been described.48 Spontaneous seizures in these animals arise nearly simultaneously in both normal and ectopic cortical circuits due to extensive interconnections between the two.16 The genes underlying this malformation have not yet been identified; however, one epileptogenic mechanism described involves a reduction in the number of parvalbumin-containing GABAergic interneurons in Tish necortex.96 This lesion is suggestive of the interneuron migratory defect seen in some targeted mouse mutants.
Absence Epilepsy in Defined Single Locus Mouse Models
Systematic electroencephalographic (EEG) screening of murine mutants for cortical excitability defects has so far revealed ten single gene loci for spike-wave epilepsy phenotypes (Table 1). All are recessive, save for the autosomal dominant Coloboma locus, a deletion containing the exocytosis-related SNAP25 gene, and many show additional neurologic phenotypes in the homozygous mutant. Except for the 3/sec spike-wave epilepsy mutant Swe lacking a functional NHE1 sodium hydrogen exchanger gene,21 these mice express spontaneous, brief (1- to 10-second), generalized 6- to 7-Hz spike-wave discharges in the cortical EEG associated with arrest of movement. Seizures in these models typically begin in the third week of life, continue into adulthood, and respond dramatically to ethosuximide. Swe mice are the sole mouse model to reproduce a 3/sec spike-wave discharge in juvenile mice, disappearing in adulthood. Neurons in NHE1-deficient mice show prolonged recovery from an acid load and increased sodium ion channel expression.104
The tottering mutant develops in adolescence (postnatal day 17) a generalized 6 to 7/sec cortical spike-wave discharge with brief (1- to 10-second) bursts occurring at a mean rate of up to 60/hr. Many laboratories have now studied neuronal
excitability in these classical calcium channel mutant models and their multiple alleles, and they confirm a range of cellular abnormalities in each. Loss of function mutations in the P/Q type α subunit of voltage-gated calcium channels reduce calcium entry through the pore; however, at many synapses, release defects are partially compensated by N-type calcium channels77 and similar compensatory rearrangements by other related subunit genes can follow the loss of channel regulatory subunits, as seen in the lethargic Cacnb4 mutant.13,14 In the tottering mouse, several key mechanisms underlie the genesis of spike-wave synchrony, including an elevation of T-type calcium currents in thalamic neurons that precedes the onset of epilepsy (also shared by lethargic, stargazer, and Coloboma models),107,108 and a reduction of feed-forward inhibition onto layer IV neurons in the neocortex,80 all correlating with the onset of epilepsy. The presence of thalamic T-type currents is essential for the expression of spike-wave discharges in these models, since deleting the Cacna1G gene encoding T-type currents in thalamic relay nuclei abolishes the seizure phenotype.44,87 Other downstream plasticity mechanisms are likely to modulate spike-wave epileptogenesis and may serve as additional therapeutic targets.70 Analysis of tottering mice also provided the first example of a model where a potential therapeutic target may be far removed from the mutant gene itself. The tottering brain shows a gene-linked proliferation of noradrenergic locus coeruleus axon terminals in neocortical and thalamic regions, and neonatal correction of this inherited hyperinnervation with a selective neurotoxin prevents the expression of epilepsy.50,65 The neuromodulatory effects of noradrenaline on the compensatory N-type calcium channel may explain why removal of this downstream target rescues the tottering phenotype.51 Lethargic and ducky mice express an absence seizure phenotype similar to tottering, including the accompaniment of paroxysmal dystonias, but since they bear mutations of regulatory subunits that interact with multiple calcium channel α subunit pore proteins (not simply Cacna1a), they share only some of the regional profile of excitability changes.
excitability in these classical calcium channel mutant models and their multiple alleles, and they confirm a range of cellular abnormalities in each. Loss of function mutations in the P/Q type α subunit of voltage-gated calcium channels reduce calcium entry through the pore; however, at many synapses, release defects are partially compensated by N-type calcium channels77 and similar compensatory rearrangements by other related subunit genes can follow the loss of channel regulatory subunits, as seen in the lethargic Cacnb4 mutant.13,14 In the tottering mouse, several key mechanisms underlie the genesis of spike-wave synchrony, including an elevation of T-type calcium currents in thalamic neurons that precedes the onset of epilepsy (also shared by lethargic, stargazer, and Coloboma models),107,108 and a reduction of feed-forward inhibition onto layer IV neurons in the neocortex,80 all correlating with the onset of epilepsy. The presence of thalamic T-type currents is essential for the expression of spike-wave discharges in these models, since deleting the Cacna1G gene encoding T-type currents in thalamic relay nuclei abolishes the seizure phenotype.44,87 Other downstream plasticity mechanisms are likely to modulate spike-wave epileptogenesis and may serve as additional therapeutic targets.70 Analysis of tottering mice also provided the first example of a model where a potential therapeutic target may be far removed from the mutant gene itself. The tottering brain shows a gene-linked proliferation of noradrenergic locus coeruleus axon terminals in neocortical and thalamic regions, and neonatal correction of this inherited hyperinnervation with a selective neurotoxin prevents the expression of epilepsy.50,65 The neuromodulatory effects of noradrenaline on the compensatory N-type calcium channel may explain why removal of this downstream target rescues the tottering phenotype.51 Lethargic and ducky mice express an absence seizure phenotype similar to tottering, including the accompaniment of paroxysmal dystonias, but since they bear mutations of regulatory subunits that interact with multiple calcium channel α subunit pore proteins (not simply Cacna1a), they share only some of the regional profile of excitability changes.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


