INTRODUCTION
Parkinson’s disease (PD) is the second most frequent neurodegenerative disorder, affecting about 2% of individuals over the age of 65. Clinically, PD patients present with the cardinal motor symptoms of bradykinesia, resting tremor, rigidity, and postural instability. Underlying these clinical features is a progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta, accompanied by an accumulation of eosinophilic intracytoplasmic protein inclusions known as Lewy bodies and Lewy neurites, both of which are the pathologic hallmarks.
For the longest time, PD was the textbook example of a “nongenetic” disorder. With the identification of several disease-causing genes, this view has changed over the last 20 years. Monogenic forms of PD are very rare and altogether account for less than 10% of PD cases. They follow the typical pattern of Mendelian inheritance with an autosomal dominant or an autosomal recessive mode with high penetrance. The disease-causing mutations are located in the coding regions of the respective genes.
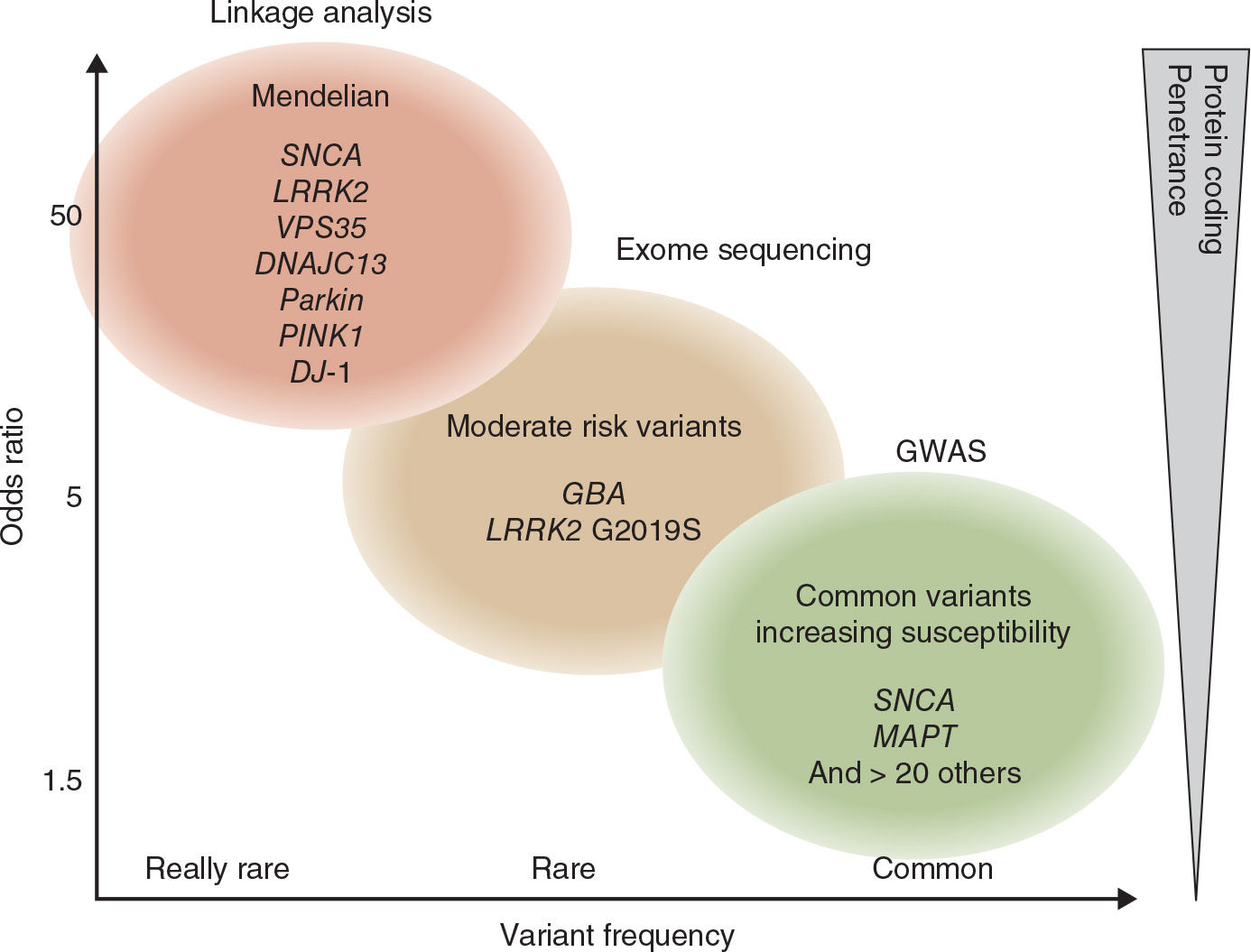
With the development of novel technologies such as genome-wide association studies (GWAS) and next-generation sequencing, it became clear that some genetic variants are not directly causative for a familial form of the disease, but rather increase susceptibility for sporadic PD. These variants are located in noncoding regions of different genes and increase the susceptibility to develop sporadic PD only by a factor of 1.5 to 2.0. Thus, they are not useful to predict an individual’s risk to develop PD, but because of their frequency, these variants explain a considerable proportion of the population-wide disease risk. For an overview, see Fig. 8.1.
Currently, a major effort is put into the evaluation whether some of these variants also act as disease modifiers and might therefore help to explain the variability of the clinical phenotype with regard to disease progression and the characteristics of different nonmotor symptoms such as dementia.
Some of the genes play a major role in several categories. They are involved in the classic Mendelian inheritance but also increase the susceptibility to develop sporadic PD and act as modifiers for the phenotype. Other genes that cause Mendelian forms of PD were reported to act in the same pathways that are also discussed in the etiology of sporadic PD and are therefore of special interest in order to elucidate the molecular pathogenesis in the sporadic form. All of them are likely to be modified by environmental factors, which, in turn, makes the analyses of the individual impact even more complicated.
MONOGENIC FORMS OF PD
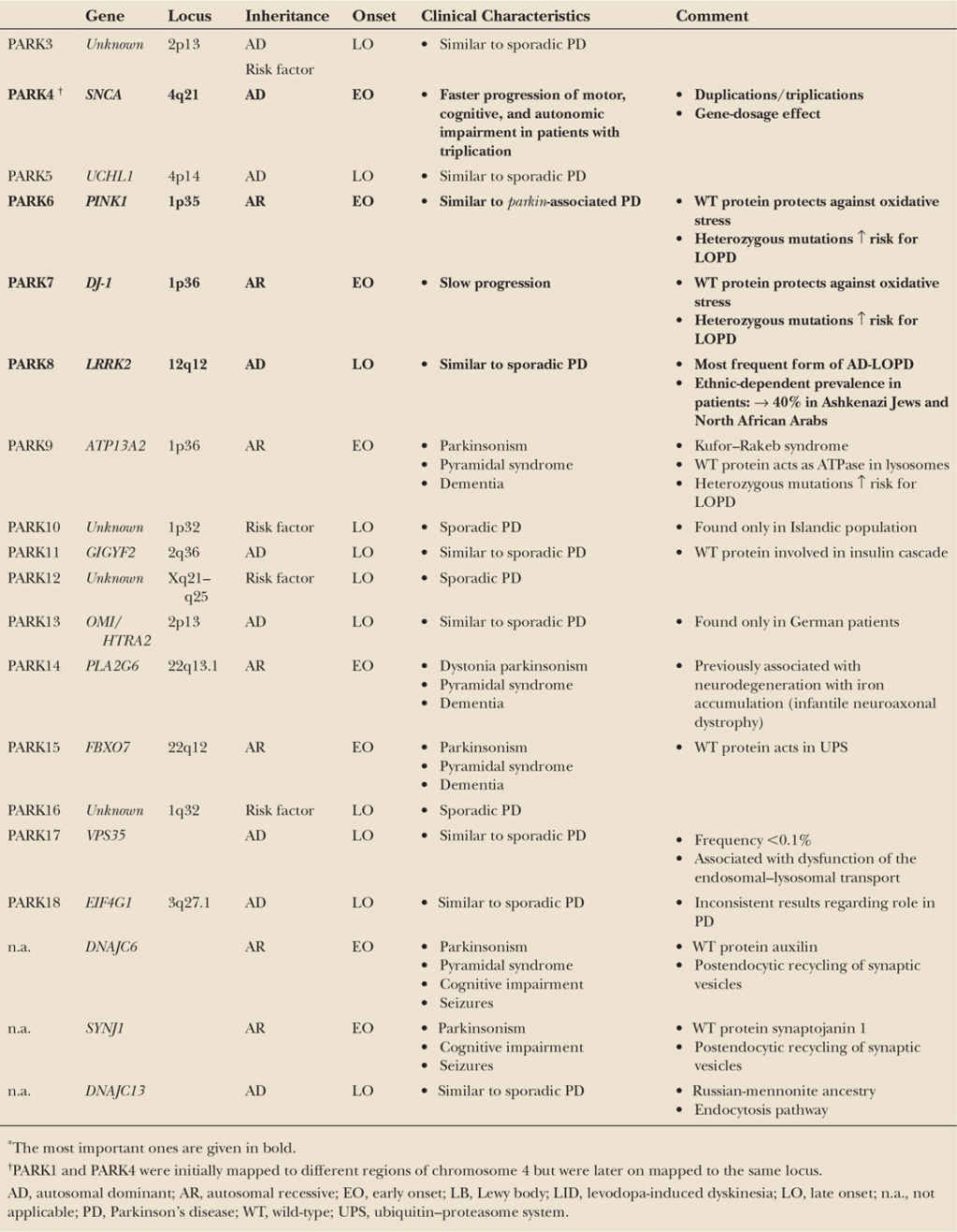
A minority of patients with the typical clinical picture of PD have a positive family history compatible with a Mendelian (autosomal dominant or autosomal recessive) inheritance. Age at motor onset in many (but not all) of these familial patients is younger than that of patients with sporadic PD, but no other specific clinical signs distinguish familial from sporadic cases. Pathologically, all forms have in common a predominant degeneration of dopaminergic neurons of the substantia nigra, although in some forms the pathologic process appears to be more selective (as in Parkin-associated parkinsonism), whereas in others the degenerative process is more widespread. Some forms demonstrate typical Lewy body pathology (SNCA), whereas in others the pathology differs from that considered to be typical for PD in some cases but not in others (LRRK2). Remarkably, pathology can vary even within single families. These observations indicate that different, and probably interrelated, pathogenic pathways are likely to lead to the process of nigral cell death. For an overview, see Table 8.1.
AUTOSOMAL DOMINANT FORMS OF PARKINSONISM
PARK1/4: PD CAUSED BY MUTATIONS IN, OR MULTIPLICATIONS OF, THE GENE FOR α-SYNUCLEIN (SNCA)
The α-synuclein (SNCA) gene was the first one found to cause familial PD. It was initially mapped to chromosome 4q21–q23 in a family of Italian origin (Contursi kindred) including more than 60 PD-affected individuals over five generations who showed an autosomal dominant pattern of inheritance due to the p.A53T missense mutation (1). Later, additional missense mutations were recognized (2,3), although p.A53T is the most frequent one and mostly found in Mediterranean families of Italian and Greek origins. Haplotype data suggest that the mutation in many of these families is based on a common founder. In general, patients carrying SNCA point mutations (PARK1) present a clinical phenotype similar to that of patients with sporadic PD. However, they have an earlier onset, and often suffer from prominent dementia, psychiatric disturbances, and autonomic dysfunction (4). In accordance with these clinical findings, autopsy revealed loss of dopaminergic neurons of the substantia nigra combined with extensive Lewy pathology, not only in the substantia nigra and other brain stem nuclei but also in the limbic system, the hippocampus, and throughout the neocortex (3,4).
Later, several groups showed that duplications and triplications of the SNCA gene can also cause autosomal dominant PD (called PARK4, because the disease locus in the first triplication family was first erroneously mapped to a different part of chromosome 4) (5,6). Hence, the involvement of SNCA in PD has been further substantiated by the fact that not only point mutations but also multiplications of the wild-type gene, leading to an overexpression of the wild-type protein, cause autosomal dominant PD. Since patients with SNCA triplications generally present with an early age of onset, severe disease progression, distinct dementia, and frequent autonomic dysfunction while patients with duplications show a more typical late-onset PD phenotype, severity of the clinical phenotype in SNCA multiplications seems to be associated with gene dosage (6).
Although SNCA mutations are rare, their identification was extremely important, as it led to the discovery that the encoded protein, α-synuclein (Asyn), is the major component of Lewy bodies (7), the proteinaceous inclusion that has, since Friedrich Lewy’s original description in 1917, been considered to be the pathologic hallmark of PD in both familial and sporadic cases. This finding supports a central role of Asyn in the development of PD. Several hypotheses on the relationship between the formation of Asyn aggregates and cell death are discussed, although the precise contribution of this protein to pathogenesis is unknown. Pathogenic amino acid changes in the Asyn protein favor the β-pleated sheet conformation, which, in turn, may lead to an increased tendency to form aggregates (8). Alternatively or in addition, failure of proteasomal degradation of Asyn and other proteins may lead to an accumulation of toxic species of the protein, possibly consisting of Asyn protofibrils (9), and ultimately leading to cell death (10). Another possible mechanism could be a deficient chaperone-mediated lysosomal clearance of Asyn (11). Recent findings demonstrate that Lewy body pathology can be experimentally transmitted by inoculating misfolded forms of Asyn, which are capable of inducing Lewy body formation through a self-propagating mechanism similar to prions. Within the brain, “seeded” Lewy bodies and Lewy neurites can also be transmitted to anatomically interconnected regions (12,13).
A relatively small protein, Asyn is abundantly expressed in many parts of the brain and localized mostly to presynaptic nerve terminals. Mice treated with the toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) revealed high mRNA and protein levels of Asyn in their brains paralleled by deposition of Asyn aggregates, whereas Asyn-deficient mice displayed resistance to MPTP-induced degeneration of dopaminergic neurons (14). Thus, pharmacologic manipulation of Asyn expression may be a possible therapeutic strategy to prevent PD in susceptible individuals.
PARK8: PD CAUSED BY MUTATIONS IN THE GENE FOR LEUCINE-RICH REPEAT KINASE 2 (LRRK2)
Another locus for a dominantly inherited form of PD has been mapped to the pericentromeric region of chromosome 12 in a large Japanese family with patients showing typical PD. Later on in 2004, point mutations in the gene for leucine-rich repeat kinase 2 (LRRK2) were identified to cause this form of dominantly inherited PD (15,16). More than 50 different missense changes have been found, although pathogenicity has been proven only for 7 mutations (p.R1441C, p.R1441G, p.R1441H, p.Y1699C, p.I2020T, p.G2019S, p.N1437S).

LRRK2-associated PD is remarkable for several reasons. Mutations in the LRRK2 gene are the most frequent genetic cause associated with autosomal dominant PD. Depending on the series, a prevalence of up to 10% in familial PD and also of 3% in sporadic PD has been found, and even up to 20% to 30% in several ethnic populations, for example Ashkenazi Jews and North African Arabs. One particular mutation (p.G2019S) should be highlighted, as it is responsible for familial PD in up to 7% of cases in different Caucasian populations, but it can also be detected in 1% to 2% of sporadic patients (17). Particularly, the high prevalence in sporadic cases suggests a reduced penetrance of this mutation. Additionally, the penetrance of p.G2019S-associated PD shows an age-dependent increase from 28% at the age of 59 to 74% at the age of 79 irrespective of the sex, which has to be taken into account for genetic counseling. Haplotype analysis implied that the p.G2019S mutation can be traced back to a few founder events in different populations as there are mainly three different haplotypes to be found in p.G2019S carriers (17).
In general, the clinical phenotype of LRRK2 mutation carriers resembles late-onset sporadic PD more closely than that associated with other PD mutations. Patients usually show a good response to levodopa treatment. However, with respect to motor and also nonmotor symptoms such as dementia and olfactory dysfunction, the clinical course in p.G2019S patients appears to be more benign than in nonmutation PD (18,19). Interestingly, homozygous LRRK2 patients as well as patients with LRRK2 mutations carrying an additional mutation within one of the PD-linked genes clinically present a phenotype similar to that of heterozygous LRRK2 patients (17). PET (positron emission tomography) studies indicate an asymmetric presynaptic dopaminergic dysfunction in the basal ganglia analogous to sporadic PD (20).
Although the clinical picture resembles typical PD, the associated pathology is remarkably variable. Pathologic changes include abnormalities consistent with typical PD-associated Lewy body pathology in most patients, but diffuse Lewy body disease, nigral degeneration without distinctive histopathology, and even progressive supranuclear palsy–like tau aggregation (15,21) have been described. Since marked variations can be observed between patients with the same mutation, clinical as well as pathologic differences do not seem to arise from the character of specific mutations. In fact, gene interactions and further modifying mechanisms or compensatory processes may influence the course of the disease.
Spanning a genomic region of 144 kb with 51 exons, the LRRK2 gene is very large and encodes a multidomain protein consisting of 2,527 amino acids. This protein belongs to the ROCO (ROC and COR domain) protein family and is made up of five characteristic functional sections named LRR domain (leucine-rich repeats), ROC domain (Ras of complex proteins), COR domain (C-terminal of Roc), MAPKKK domain (mitogen-activated protein kinase kinase kinase), and WD40 domain (15). Each domain seems to be involved in specific functions, and therefore, different mutations could interfere with different tasks. The p.G2019S and p.I2020T mutations are located within the kinase domain, and in vitro studies indicated that these mutations may confer a “gain of function mechanism” resulting in an increased (auto)phosphorylation (22). Thus, specific kinase inhibitors are discussed as potential therapeutic strategy. In addition, LRRK2 is a GTP/GDP-regulated protein kinase, combining both the kinase effector and the GTP-binding regulatory domain (ROC domain) within one protein. Mutations within the ROC domain may therefore be able to influence the binding activity of the kinase domain (23). Molecular pathways implicated in LRRK2 function in neurons include maintenance of the actin skeleton (24), recycling of the presynaptic vesicle pool (25), or the clearance of Golgi-derived vesicles through the autophagy-lysosome system (26). Recent findings support a role of LRRK2 in the immune system, suggesting inflammatory pathways in LRRK2-associated PD and thereby offering potentially new therapeutic windows (27).
PARK17: PD CAUSED BY MUTATIONS IN THE GENE FOR VACUOLAR PROTEIN SORTING 35 (VPS35)
In 2011, two groups reported the p.A620A mutation in the VPS35 gene as a novel cause of autosomal dominant PD (28,29). Gene identification was facilitated by next-generation sequencing technology, specifically “whole exome sequencing,” which allows to rapidly identify thousands of missense variants in two or more affected individuals of large families, which can then be studied for cosegregation. Subsequent studies could support this specific mutation as a rare disease-causing variant in Caucasian and Japanese familial PD patients with a frequency of <1%. The variant was found in 10 of 7,989 cases, but in only 1 of 6,513 controls (30). While this difference is statistically significant, the study also shows that very large sample sizes are necessary to confirm pathogenicity of rare disease variants. The phenotype resembles that of typical late-onset PD. The encoded protein is a component of the so-called retromer complex and is involved in endosomal trafficking and recycling of synaptic vesicles.
PD CAUSED BY MUTATIONS IN THE DNAJC13 GENE
Recently, mutations in the DNAJC13 gene were found to cause autosomal dominant PD in patients of Russian-Mennonite ancestry (31). Postmortem examination shows typical Lewy body pathology. The encoded protein is involved in the endocytosis pathway, which strengthens the link between endocytosis, retrograde axonal transport, Lewy body formation, and PD.
AUTOSOMAL RECESSIVE FORMS OF PARKINSONISM
An important achievement was the identification of several genes causing recessively inherited early-onset PD. To date, three major genes have been identified: Parkin (PARK2), PINK1 (PARK6), and DJ-1 (PARK7). Studies on the function of these genes have provided valuable insight into the molecular mechanisms of dopaminergic degeneration.
PARK2: AUTOSOMAL RECESSIVE JUVENILE PD CAUSED BY MUTATIONS IN THE GENE FOR PARKIN (PRKN)
The PARK2 locus, spanning a large region on chromosome 6, was the first locus to be linked to an early-onset form of PD following an autosomal recessive transmission. Initially mapped in several Japanese families, positional cloning identified homozygous deletions in a large gene in this region that was named parkin (PRKN) (32). Subsequently, various types of mutations (deletions, multiplications, point mutations) within the parkin gene were identified, and it soon became clear that these mutations are a frequent cause of autosomal recessive juvenile PD. Among families with early-onset PD under the age of 30, 49% of the patients with an affected sibling had parkin mutations as opposed to about 10% of cases without positive family history. There was a clear age dependency of the prevalence of parkin mutations (82% with an onset before age 20 years) (33).
In parkin mutation carriers, age at onset is usually before 40 years and ranges from the second to the fourth decade. In the beginning, dystonia is a common feature, sometimes raising difficulties in distinction from dopa-responsive dystonia. Over time however, most patients with parkin mutations develop early and marked levodopa-induced motor fluctuations and dyskinesias, although the disease seems to progress only slowly with a low prevalence of dementia. Independent of the underlying type of mutation (deletions, multiplications, point mutations), patients demonstrate the full phenotype of early-onset parkinsonism, indicating that a complete loss of parkin function is associated with all of these mutations (33).
Corresponding to the rather symmetric motor impairment observed clinically, functional neuroimaging with PET also demonstrated a symmetrically reduced striatal uptake of the tracer in both hemispheres, more pronounced in the putamen than in the caudate nucleus (34). This in fact differs from the initially unilateral reduction of (18F)fluorodopa uptake in PD patients without parkin mutations.
Whether heterozygous parkin mutations increase the susceptibility to late-onset PD is widely discussed. The observation that heterozygous mutation carriers in families with homozygous relatives have been found to present with minor signs of basal ganglia dysfunction or late-onset parkinsonism has been interpreted as evidence that a single mutant allele acts as a susceptibility factor, increasing the risk of developing PD (35). However, some groups reported comparable frequencies of parkin variants in PD cohorts and controls (36,37), arguing against a pathogenic role of heterozygous mutations.
Histopathologic studies in parkin-related cases revealed severe neuronal degeneration and gliosis in the substantia nigra and moderate decrease of neurons in the locus coeruleus, while Lewy bodies have not been described in most cases (38). However, two cases with compound heterozygous mutations showed Lewy bodies at autopsy (39), indicating that Asyn aggregation is a possible but not obligatory feature of parkin-related parkinsonism.
Since the parkin protein acts as E3-ubiquitin ligase, it has initially been linked to the ubiquitin–proteasome system which is mainly involved in protein degradation. It was assumed that through a “loss of function” due to a mutation in the parkin gene, the impaired E3-ubiquitin ligase activity would lead to the accumulation of parkin-dependent substrates and thus to nigral degeneration (14). However, despite many initial promising leads, no such accumulating substrate has ever been confirmed.
Today, the leading hypothesis states that parkin mutations are pathogenic by impairing mitochondrial function. For example, parkin protects mitochondrial DNA from oxidative damage and stimulates its repair. This ability is impaired by pathogenic parkin point mutations. Moreover, parkin-deleted fibroblasts of PD patients present a higher susceptibility of mitochondrial DNA to reactive oxygen species and reduced mitochondrial DNA repair capacity (40).
The most convincing line of evidence today links parkin and PINK1 to the process of elimination of damaged mitochondria by a process called mitophagy. It has been shown in several experimental systems that cytoplasmic parkin is recruited to damaged mitochondria, where it is phosphorylated by the mitochondrial kinase PINK1 (see below), and subsequently ubiquitinates other mitochondrial membrane proteins, thereby promoting the elimination of dysfunctional mitochondria through mitophagy. Since damaged mitochondria produce increased amounts of toxic oxygen radicals, a failure of their clearance results in enhanced cellular damage.
Moreover, there is growing evidence that parkin is also linked to other PD-causing pathways. Rat models showed that Asyn-induced dopaminergic neuronal cell death is rescued by overexpression of parkin, indicating a potential neuroprotective role of parkin in the pathogenesis of PD (41). This finding was further supported when LRRK2-mutant drosophila flies exhibiting loss of dopaminergic neurons, combined with motor impairment and increased sensitivity to rotenone, a mitochondrial complex I inhibitor, were protected against dopaminergic neurodegeneration by coexpression of parkin (42).
PARK6: AUTOSOMAL RECESSIVE JUVENILE PD CAUSED BY MUTATIONS IN THE GENE FOR PINK1 (PINK1)
In a large Sicilian family with autosomal recessive early-onset PD, Valente and colleagues identified a novel locus, PARK6, mapped to chromosome 1p36–p35 (43). Subsequently, additional unrelated families showed positive linkage to this region, and several novel mutations were found, indicating that PINK1 mutations might be the second most common cause for familial early-onset PD.
Except the early onset around the thirties and slow disease progression, the clinical phenotype resembles typical PD presenting unilateral beginning, resting tremor, bradykinesia, and rigidity with good response to levodopa. However, an extended phenotype with psychiatric impairment, including drug-induced behavioral disturbances and psychosis as well as frontal-type dementia, was also reported (44). Again, some studies imply heterozygous mutations to contribute to the development of late-onset PD (35).
Typical pathologic hallmarks of sporadic PD, including nigral neurodegeneration combined with Lewy bodies in the brain stem and neocortex, were reported in a compound heterozygous patient with early onset and in four patients with late onset carrying heterozygous mutations in the PINK1 gene (45,46). This was surprising, as PINK1 and parkin are generally thought to be closely related with respect to their pathogenic mechanisms, and Lewy bodies are generally not found in parkin cases.
In human brain, PINK1 is widely expressed and localizes to mitochondria, especially to the intermembrane space (46). PINK1 contains an N-terminal mitochondrial-targeting signal and a catalytic kinase domain. Although specific functions of PINK1 still remain unclear, in vitro experiments suggest that it might provide protection against oxidative stress-induced cell death. TNF receptor-associated protein 1 (TRAP1), a mitochondrial chaperone, was identified as a substrate for PINK1. Through phosphorylation of TRAP1, PINK1 protects against oxidative-stress-induced cell death by suppressing cytochrome c release from mitochondria. The ability of PINK1 to enhance TRAP1 phosphorylation is affected by PD-associated PINK1 mutations (47). The protective role of PINK1 was further supported by the observation that PINK1-null mice revealed alterations of dopamine release at synaptic terminals and impaired mitochondrial function (14).
As described earlier, there is growing evidence that PINK1 and parkin act in a common pathway to eliminate damaged mitochondria by mitophagy. Interestingly, the degeneration of dopaminergic cells and the mitochondrial phenotype in PINK1-deficient drosophila could be rescued by overexpression of parkin (48). These findings are of particular interest since mitochondrial dysfunction is discussed as a possible factor in the etiology of sporadic PD.
PARK7: AUTOSOMAL RECESSIVE JUVENILE PD CAUSED BY MUTATIONS IN THE GENE FOR DJ-1 (DJ-1)
Mutations in the DJ-1 gene linked to chromosome 1p36 represent a rare cause for autosomal recessive early-onset PD with a frequency of approximately 1% (49). Over the years, homozygous missense mutations, compound heterozygous mutations, as well as deletions were identified.
The clinical phenotype is similar to that described in patients with parkin and PINK1 mutations.
DJ-1 is ubiquitously expressed and, in human brain, particularly localized to astrocytes, whereas DJ-1 immunoreactivity in neurons is only weak (50). DJ-1 is not a characteristic component of Lewy bodies but was found in glial inclusions and neuronal tau inclusions in α-synucleinopathies and tauopathies (51).
The function of DJ-1 is not entirely clear, but it is postulated to act as a redox-responsive cytoprotective protein in an antioxidant manner. DJ-1-knockout drosophila displays cellular accumulation of reactive oxygen species combined with hypersensitivity to oxidative stress and degeneration of dopaminergic neurons (14). Moreover, DJ-1 is upregulated in reactive astrocytes in chronic neurodegenerative diseases as well as in stroke, acting as redox-reactive pointer controlling oxidative stress after ischemia, upon neuroinflammation, and during neurodegenerative processes (52).
AUTOSOMAL RECESSIVE PARKINSONISM CAUSED BY MUTATIONS IN THE GENES DNAJC6 AND SYNJ1
During the past year, mutations in these two genes were identified as the cause of autosomal recessive juvenile parkinsonism with an atypical phenotype including cognitive impairment and seizures. The encoded proteins (auxilin, synaptojanin 1) are involved in the postendocytotic recycling of synaptic vesicles (53,54).
UNCONFIRMED GENES AND LOCI THAT MIGHT BE ASSOCIATED WITH PD
The pathogenicity of several loci and genes that have been suggested by linkage analysis or a candidate gene approach and were originally associated with PD could not so far be unequivocally confirmed as disease genes. Some of these genes even attained a “PARK” designation (PARK3, UCHL1-PARK5, PARK10, GYGYF2-PARK11, OMI/HTRA2-PARK13, EIF4G1-PARK18). The link of these genes to PD is uncertain. Furthermore, mutations in some PARK loci cause rare syndromes of a more complex phenotype, with parkinsonism representing only one of several clinical features (ATP13A2-PARK9, PLA2G6-PARK14, and FXBOX – PARK15). For an overview, see Table 8.1.
GENETIC CONTRIBUTION TO SPORADIC PD
Although genetic analyses have produced significant progress in families with Mendelian inheritance, it must be remembered that in the great majority of cases PD is a sporadic disorder. However, the fact that genetic factors also play a role in families without a clear inheritance pattern is now firmly established. Cases of PD are found more frequently among relatives of patients with PD than in the general population. Population-based case–control studies indicate that the relative risk for first-degree family members of PD patients is increased in the order of 2 to 3 (55,56).
With advancing technologies, new approaches toward the identification of putative genetic risk factors for sporadic PD became available. Only recently, GWAS assessing large case–control populations including thousands of individuals identified common sequence variations in the genes for α-synuclein (SNCA) and microtubule-associated protein tau (MAPT) as the first risk loci for sporadic PD (57). These variants are generally located not in the coding region of the genes, but in introns, promotor regions, or 5′ untranslated parts of the gene, most likely affecting the regulation of gene transcription. They are not directly disease-causing but convey a mildly (usually by a factor of 1.1–2) increased risk for the development of PD. Although individually weak, owing to their common occurrence they may explain up to 10% of the disease risk of the whole PD population. In the last 5 years, additional risk loci were reported (58). Interestingly, the identification of the HLA-DR gene as one of those risk loci supports the hypothesis of an immunologic impact in the pathogenesis of PD. For an overview, see Table 8.2.
Remarkably, one of the most important genetic risk factors for sporadic PD was missed by the new high-throughput genotyping and GWAS technologies. The observation that mutations in the glucocerebrosidase (GBA) gene are a genetic risk factor for PD came into focus as a result of astute clinical observations of patients with Gaucher’s disease (GD) and their families. GD is the most common lysosomal storage disorder with tissue accumulation of glucocerebrosides due to deficiency of the enzyme glucocerebrosidase (GCase) on the basis of homozygous mutations in the GBA gene. From the first descriptions of PD and GD in the 19th century, it took more than 100 years to discover the link between the GBA gene and parkinsonism. About 25% of relatives of GD patients who were obligate or confirmed heterozygous GBA mutation carriers demonstrated typical parkinsonism (72,73). This finding was the important hint that heterozygous mutations in the GBA gene might be associated with PD and led to subsequent mutational screening in patient cohorts with sporadic PD. In 2009, a large multicenter analysis with 16 participating centers from four continents compared 5,691 PD patients of different ethnic origin with 4,898 control individuals without PD. All subjects were screened for at least the two most common GBA mutations (p.L4444P, p.N370S) which were found in 15% of Ashkenazi Jewish PD patients and in 3% of Ashkenazi Jewish control individuals as well as in 3% of PD patients of other ethnic origin and in less than 1% of non-Ashkenazi Jewish controls. The study confirmed that with an overall OR of 5.43, heterozygous mutations in the GBA gene represent the most common genetic risk factor for PD so far (64).
Clinically, PD patients carrying a GBA mutation present with an earlier age at onset and more frequently suffer from nonmotor symptoms such as cognitive impairment, neuropsychiatric disturbances, and autonomic dysfunction, compared with PD patients without GBA mutations (74). Moreover, the mutational GBA status predisposes to a more rapid disease progression of motor impairment and cognitive decline and is associated with reduced survival rates (75).
| Overview of Genetic Risk Factors Associated with Sporadic Parkinson’s Disease and Its Impact on the Development of the Disease* |