CHAPTER 141 Glomus Tumors
The original oncotype is considered to be the “glomus cell,” a modified neuroblast of the neural crest; it differentiates into an epithelioid type that migrates during embryogenesis toward the autonomic ganglia1 and has the potential to store and secrete catecholamines in response to neuronal or chemical signals.2–4 The largest collection of these “neurocrine” cells is found in the adrenal medulla. Smaller colonies are physiologically present in the cardioaortic and laryngeal bodies near the carotid bifurcation, the adventitia of the jugular bulb, the promontory of the middle ear, and the vagal nerve, but their neoplastic representatives may be found in a wider range of locations.5–22
As a result of the various locations, a variety of eponyms have been coined to characterize this oncologic lineage. The term chemodectoma has mostly been used to refer to carotid body tumors because of their oxygen sensor,1,23,24 whereas the term paraganglioma or nonchromaffin paraganglioma and, in particular, glomus tumor or glomangioma have arbitrarily been used when dealing with other anatomic sites more familiar to neurosurgeons, otolaryngologists, and head and neck surgeons. Specifically, these sites include (1) the jugular bulb and foramen (“glomus jugulare”); (2) Jacobsons’s nerve, or the tympanic branch of the glossopharyngeal nerve (“glomus tympanicum”); and (3) Arnold’s nerve, or the auricular branch of the vagus nerve (“glomus vagale” or “juxtaovale”).23,25 Finally, the acronym “APUDOMA”26 or “intracranial pheochromocytoma”3 was created to define the limited fraction of secreting, non–chromaffin-containing paragangliomas that—similar to carcinoid tumors—maintain the potential for Amine Precursor Uptake and Decarboxylation, sometimes associated with carcinoid syndrome.27
Historical Background
The carotid body was first described by Van Haller in 1743,28 whereas the first surgical removal of the carotid body was performed by Riegner in 1880.29 The earliest reports on glomus tympanicum date back to Valentin (1840), who thought that the small cellular cluster near the tympanic nerve was a ganglion,30 and to Krause (1878), who was somehow impressed by the similarity between the glomus and the carotid body.31 In 1945, Rosenwasser, reporting on the surgical removal of a “carotid-body-like tumor” located in the middle ear, further stressed this consistent analogy.32 In 1941, Guild described the glomus jugulare in the adventitia of the jugular bulb.33
A few years later (1951), neoplastic lesions arising from paraganglionic cells in this area were called glomus jugulare tumour by Winship and Louzan.34 Finally, in 1953, Guild extensively analyzed a large series (88 specimens) of human temporal bone sections and substantially outlined the current glomus nosography by classifying the three major varieties as jugulare, tympanicum, and vagale.35
Surgical exploration of the jugular bulb was bravely initiated by Seiffert in 1934, Lundgren in 1949,36 and more skeptically by Weille and Lane in 1951.37
The suboccipital approach to this region was first reported in 1953 by Semmes38 and by Albernaz and Buzcy.39 During the following 2 decades, more aggressive surgical techniques were gradually introduced with the aim of removing even larger tumors usually considered inoperable, as well as to gain complete “anatomic control” of such a crucial area. The main achievements in this period, which were put into practice by Shapiro and Neues in 196440 and Gejrot in 1965,41 led to progressively more extended craniocervical incisions and wider skull base approaches, including either established techniques, such as labyrinthectomy (Gastpar, 196142) and radical mastoidectomy (Hilding and Greenberg, 197143), or sophisticated refinements such as mobilization and rerouting of the facial nerve for packing/resecting the sigmoid sinus or jugular bulb40–43 and eventually to temporal bilateral carotid artery occlusion with hypothermia.44 In 1969, House proposed his famous approach through the facial recess, which resulted in higher rates of cranial nerve preservation.45 This outstanding innovation was followed in the late seventies and eighties by the infratemporal fossa route proposed by Fisch and coworkers to overcome problems with exposure of the internal carotid artery46–48; the major variations of this technique were conceived to tackle a spectrum of anatomic extensions and different neoplasms of medium and large size.
In contrast, more limited approaches were devised for safe removal of smaller lesions: Mischke and Balkany’s “posterior-inferior” (1980) technique for sparing both the auditory and facial canal49 and Farrior’s (1984) “anterior hypotympanic” technique.50
In the mid-1980s, Brown published one of the largest surgical series,51 Donald and Chole described their transcervical-transmastoid approach,52 and above all, Gardner, Robertson, and colleagues reported their wide experience46,53; the latter repeatedly stressed the importance of comprehensive team management of these tumors and advocated cooperative effort by otologists, neurosurgeons, craniofacial surgeons, anesthesiologists, internal medicine specialists, and rehabilitation experts not only to optimize surgical performances but also to improve clinical and neurological outcomes. Their famous “lateral skull base approach” is a prime example of this multidisciplinary attitude.4,23,46,53
A wide spectrum of different grading systems were gradually introduced into the literature: Kinney54; Fisch (Table 141-1)47; Jackson, Glasscock, and coworkers (Table 141-2)55; Rockley and Hawke56 and Alford and Gilford57 (Table 141-3) (Figs. 141-1 and 141-2); and Spector and colleagues (Table 141-4).58
TABLE 141-1 Fisch Classification of Glomus Jugulare Tumors
| CLASS | DESCRIPTION |
|---|---|
| A | Tumor limited to the middle ear cleft (glomus tympanicum) |
| B | Tumor limited to the tympanomastoid area with no involvement of the infralabyrinthine compartment |
| C | Tumor involving the infralabyrinthine compartment of the temporal bone and extending into the petrous apex |
| C1 | Tumor with limited involvement of the vertical portion of the carotid canal |
| C2 | Tumor invading the vertical portion of the carotid canal |
| C3 | Tumor invading the horizontal portion of the carotid canal |
| D | |
| D1 | Tumor with intracranial extension <2 cm in diameter |
| D2 | Tumor with intracranial extension >2 cm in diameter |
From Fisch U. The infratemporal fossa approach for extensive tumors of the temporal bone and base of the skull. In: Silverstein H, Norrell H, eds. Neurological Surgery of the Ear. Birmingham, AL: Aesculapius; 1977:34-53.
TABLE 141-2 Glasscock-Jackson Classification of Glomus Jugulare Tumors
| CLASS | DESCRIPTION |
|---|---|
| I | Small tumor involving the jugular bulb, middle ear, and mastoid |
| II | Tumor extending under the internal auditory canal; may extend into the intracranial canal |
| III | Tumor extending into the petrous apex; may extend into the intracranial canal |
| IV | Tumor extending beyond the petrous apex into the clivus or infratemporal fossa; may extend into the intracranial canal |
From Jackson CG, Glasscock ME 3rd, Nissen AJ, et al. Glomus tumor surgery: the approach, results, and problems. Otolaryngol Clin North Am. 1982;15:897-916.
TABLE 141-3 Synoptic Comparison of “Predictive” Gradings in the Rockley-Hawke56 (1990) and Alford-Gilford57 (1962) Categorization
| ROCKLEY-HAWKE | ALFORD-GILFORDPrognosis | |
|---|---|---|
| Type of Glomus Tumor | Arising From | |
| Glomus tympanicum | Good | |
| Glomus jugulare | Poor | |
| Other | Variable | |
Data from Rockley TJ, Hawke M. Glomus bodies in the temporal bone. J Otolaryngol. 1990;19:50-56; and Alford BR, Gilford FR. A comprebensive study of tumors of the glomus jugulare. Laryngoscope. 1962;72:765-805.
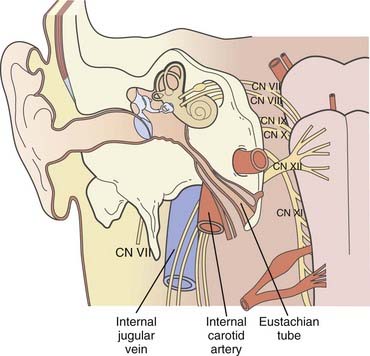
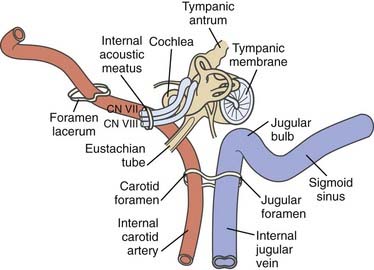
TABLE 141-4 Classification of Glomus Jugulare Tumor Spreading Patterns According to Spector and Colleagues
| CLASS | DESCRIPTION |
|---|---|
| I | Down the eustachian tube to the nasopharynx and foramina of the skull base |
| II | Along the carotid artery into the middle fossa |
| III | Along the jugular vein into the posterior fossa |
| IV | Through the tegmen tympani to the floor of the middle fossa |
| V | Through the round window of the labyrinth, invading the internal auditory canal, into the cerebellopontine angle |
From Spector GJ, Druck NS, Gado M. Neurologic manifestations of glomus tumors in the head and neck. Arch Neurol. 1976;33:270-274.
Despite their discrepancies in terms of specific coordinates, these grading systems have been extremely useful in planning surgical strategies and have been shown to reliably predict clinical outcomes. The use of more extensive, combined infratemporal and posterior fossa approaches to achieve adequate exposure, even with large lesions, as well as to avoid multiple-stage surgery, has repeatedly been emphasized in the last 2 decades. Al-Mefty and colleagues in 1987,59 Bordi and coworkers in 1989,60 Patel and associates in 1994,61 and Al-Mefty and Teixeira in 200262 have exhaustively shown the potential of integrated surgical routes (subtemporal/infratemporal/retrosigmoid–extreme lateral–transcondylar, and so on), particularly when dealing with the specifically defined “complex” variety of GTs,62 such as giant, multiple, secreting, or malignant lesions that have previously failed endovascular or radiation treatment.
A major advance in surgical results is certainly related to the introduction of preoperative superselective embolization, with Hilal and Michelsen in 197563 and Brismar and Cronqvist in 197864 providing the original “basic” contributions and Simpson and coworkers publishing their experience with GT in 1979.65 Given the limited tumor volume coverage of current techniques and the significant rates of revascularization after endovascular occlusion, embolization per se represents a palliative option that is often effective in reducing tinnitus, vertigo, and dizziness.66,67 As confirmed by accurate analyses, this technique is essentially “ancillary” to surgery and provides a substantial reduction in both intraoperative blood loss and operating time while increasing the extent of surgical resection.64–6668 However, endovascular procedures do not seem to reduce clinical sequelae; in fact, they may entail additional neurological risk.25,68–70
Fractionated radiation therapy (FRT) was first proposed as an alternative to surgery for critical GTs in 1952: Capps treated four consecutive patients and achieved good results in all of them.70 Subsequently, “conventional” radiotherapy has been used for GTs either as a primary or salvage treatment modality.71 Mainly because of their documented dose-response correlation,72,73 GTs may be considered relatively radiosensitive oncotypes,74,75 although this concept remained controversial for decades after a skeptical analysis published by Spector and associates in 197376 and the ensuing debate on parameterized radiation dose/effect with these tumors.75,77
Stereotactic radiosurgery (SRS) has recently been proposed as a potential therapeutic option for GTs.77–82 Because of the particular conformity and selectivity of updated radiosurgical dose planning in these tumors, SRS may achieve local tumor control (LTC) rates and clinical responses equivalent to or even higher than those obtained with FRT, and the incidence of sequelae seems to be lower because of the smaller volume of normal tissue exposed to radiation.77,78,81,82
Epidemiology
GTs are rare, with an incidence of approximately 1.2 to 3 cases per million people77,84; a strict minority are genetically inherited.85–88 GTs account for 0.012% to 0.03% of all newly diagnosed neoplasms and 0.6% of all head and neck tumors.78,89–91 Clinical onset is typically in the sixth and seventh decades of life,51,77,78,80,81,92,93 with a marked predilection in women and a female-to-male ratio as high as 5 : 1 to 6 : 1.51,77,78,80,81,89,92–95 Furthermore, the incidence of GTs is higher in populations living at higher altitudes than in people living at sea level96 and in patients suffering from hypoxemic conditions such as chronic obstructive pulmonary disease.96–98
In the statistically predominant nonfamilial tumors, 10% of cases are multicentric.85–86 In the 20% to 40% familial forms, bilateral involvement is rather common,87,88 and multicentricity occurs in 25% to 78% of patients.89,99,100 The tumor development rate seems consistent with autosomal dominant inheritance.89,99–101
Clinical Signs and Symptoms
The vast majority of GTs are indolent, with slow, locally invasive and destructive growth. The estimated average interval between the first symptom and diagnosis is 4 to 6 years,75,77 and the median doubling time has been calculated to be 4.2 years.89 Neurological manifestations are primarily related to the specific “region” infiltrated, which may eventually involve neurovascular structures within the temporal bone, particularly the middle ear; distal structures such as the jugular foramen, hypoglossal canal, and clivus; and more cephalad, the intrapetrous carotid artery and cavernous sinus.92,102 GTs can quite rarely become an immediately life-threatening condition, but in the mid- to long-term range, they usually jeopardize quality of life.103
In the typical glomus tympanicum tumor the basic complaints at onset are mostly pulsating tinnitus, followed by conductive hearing loss and vertigo.23,104–106 On otomicroscopic examination, the lesion appears reddish to bluish beneath the eardrum (Fig. 141-3).
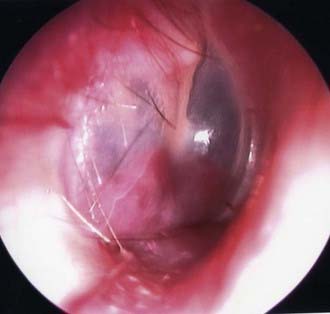
The tympanic membrane can be perforated by neoplastic invasion of the external auditory canal, and bloody otorrhea may occur. Involvement of the lower cranial nerves is usually the next step in the tympanic variety and a more precocious feature in the jugulare or vagale type. According to the literature, facial nerve dysfunction affects 8% to 33% of patients,107,108 paresis of the vagal nerve affects 6% to 30%,109 and dysfunction of the glossopharyngeal, spinal accessory, and hypoglossal nerves occurs less frequently.110,111
GTs usually have a nonsecreting phenotype.68,92,102,112,113 The approximately 1% to 3% catecholamine-secreting histotypes101,114 seem to be more common in males and younger patients.112 In such cases, the initial symptoms may include a serotonin hypersecretion–related carcinoid syndrome4,23,27 consisting of headache, facial flushing, mild hypertension, and diarrhea.23,27
A strict minority of these tumors may behave quite aggressively. Malignant variants account for 2% to 4.5% of all reported cases,57,93,115–117 with the preferred sites of colonization being the lymph nodes, liver, and lung.93,110,116–118
Pathology
However, several different anatomic-surgical classifications have been proposed. Because most of them are based either on controversial histogenetic issues or on the original extension and spreading pathways of the tumor (i.e., tumor growth topography), this particular nosography has frequently been used for predicting the natural history and surgical outcome. The preliminary study of Alford and Gilford (1962) was the first to differentiate the relatively more benign course of the tympanic form from the worse profile of the jugular variety.57 The concept was further stressed by Kinney,54 who devised one of the first staging systems for GTs based on intracranial extension, and by Rockley and Hawke in 1990, who based the prognosis of the three major varieties on the original site of the lesion.56
As mentioned earlier, Fisch47 (1977) and Jackson and colleagues55 (1982) exhaustively defined the main surgical-prognostic categories of GTs according to the size and location of the lesion, and their work received wide clinical and scientific consensus. This idea had been heralded by Spector and colleagues (1976)58 with their original classification of the main anatomic spreading pathways of these tumors (see Table 141-4). The degree of bony involvement was the crucial cutoff point for predictability.
Histology
Glomus and carotid body tumors share common histologic patterns. The dominant oncotypes are large, polygonal epithelioid cells with scanty, eosinophilic, fine granular cytoplasm that may be positive for norepinephrine on immunohistochemistry. The rounded, hyperchromatic nuclei may sometimes have mitotic figures, as well as prominent nucleoli or other atypical features (Figs. 141-4 and 141-5). However, these cytologic hints of malignancy, easily trackable in most of these specimens, poorly correlate with the effective metastatic potential of the tumor, which is rarely displayed.4,23,89,117
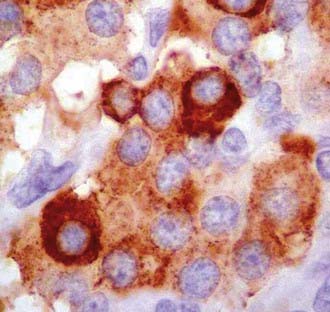
FIGURE 141-4 Typical oncotypes: large polygonal cells with scant eosinophilic, fine granular (chromogranin) cytoplasm.
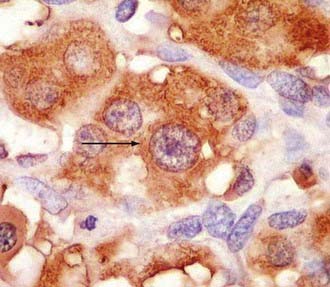
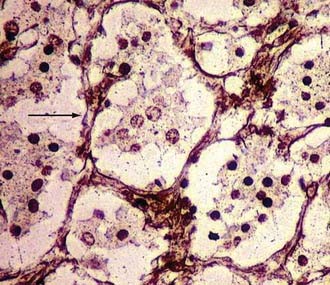
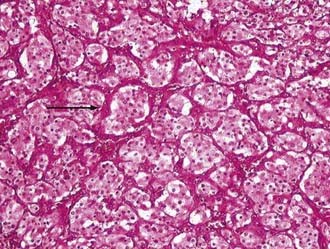
As regards histoarchitecture (Figs. 141-6 and 141-7), these cells usually form clusters or nests, hence the term Zellballen, interspersed among a reticulin network full of thin-walled capillaries. These capillaries provide an unusually rich blood supply that attracts vessels not only from the external carotid system but also from branches of the internal carotid artery.
Genetics and Molecular Biology
Familial GTs represent approximately 20% to 40% of reported cases, and the genetic defects are known just partially.24 In the largest subgroup (10% of all GTs), the cause has been documented to be sporadic germline mutations in two putative regions on chromosome 11: PGL1 at 11q23.1 and PGL2 at 11q13.1.119–122 Both regions encode for succinate dehydrogenase (SDH) subunits, the former for the putatively most relevant subunit “D,” which is localized in the mitochondrial membrane and participates in the Krebs cycle and the mitochondrial respiratory chain. Baysal and coauthors proposed that SDH is a critical component of a cellular oxygen-sensing system.123 Mutations in SDH may incapacitate this oxygen relay and lead to a hypoxic state that triggers cell proliferation. Support for this hypoxia-induced hyperplasia comes, as seen earlier, from epidemiologic studies showing an increased incidence of GTs in people living at higher altitude or suffering from chronic respiratory diseases.96–98124
An additional locus, PGL3, has been identified on chromosome 1q.125,126 Reliable genetic linkage explaining the observed association with neurofibromatosis has never been found.127 The pathogenesis of nonfamilial forms remains unexplained, but the most attractive theories involve angiogenesis mechanisms and apoptosis. Most angiogenesis-related molecules (fibroblast growth factor, platelet-derived growth factor, transforming growth factor-β1, and so on) are poorly represented in these tumors. In contrast, vascular endothelial growth factor (VEGF) and platelet-derived endothelial cell growth factor are extensively represented at the cellular (65%) and stromal (77%) levels, respectively.128 Similar observations have been reported for endothelin-1.129 Experimental protocols have shown that anti-VEGF treatment may reduce GT size in rats.130
Finally, with regard to apoptosis, high bcl-2 expression levels have been found in GTs.131 Because bcl-2 is an antiapoptotic homologue of bax and overexpression of it forestalls apoptosis and thus increases cell survival, this molecule, like murine double minute oncogene (MDM2), nuclear factor NF-κB,132 and other substances inhibiting apoptotic cascades, is currently being investigated to explore its role in the indolent course of these tumors.131
Diagnosis
In selected cases, a potential role of fine-needle aspiration biopsy is still recognized.133,134 However, to date, the basic diagnostic steps in these patients have essentially involved neuroradiology.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


