, Cenk Sokmensuer2 and Ece Esin3
(1)
Department of Medical Oncology, Cancer Institute, Hacettepe University, Sihhiye, Ankara, Turkey
(2)
Department of Pathology, School of Medicine, Hacettepe University, Ankara, Turkey
(3)
Department of Medical Oncology, Hacettepe University Institute of Cancer, Ankara, Turkey
15.1 Introduction
Glucagonomas are neuroendocrine tumours of the pancreas (pNET) originating from alpha islet cells, which synthesize and secrete glucagon or other peptides derived from the preproglucagon gene. These tumours are associated with a well-described glucagonoma syndrome characterized by hyperglucagonemia, skin rash, glucose intolerance, hypoalbuminemia, weight loss, and anemia [2].
15.2 History
The glucagonoma syndrome was first reported by Becker et al. in a patient with skin rash associated with pancreatic tumour in 1942 [3]. However, the syndrome initially was not attributed to hypersecretion of glucagon until McGavran et al. reported the first case of a patient with the glucagonoma syndrome, who had elevated plasma immunoreactive glucagon level, diabetes mellitus, skin rash, and pNET in 1966 [1]. In 1974, Mallinson et al. reported the association of skin rash with hyperglucagonemia, describing that nine cases with clinical glucagonoma syndrome consisted of dermatitis, diabetes mellitus, unexplained weight loss, hypoaminoacidemia, anemia, and a glucagon-producing tumour of the pancreas [4]. In 1984, Wilkinson described necrolytic migratory erythema (NME) for the skin rash associated with pancreatic tumours [5].
15.3 Epidemiology
Representing 1 % of all pancreatic NETs, the incidence of glucagonoma is about 1 in 20 million [6]. Patients usually present between the ages of 19 and 72 years, typically around 50’s. No glucagonoma case has been reported in children yet [7]. Although initially a female tendency was suggested in the incidence [6], in the most recent series, an even distribution among men and women has been reported [7]. No race predilection is known for glucagonoma either.
15.4 Molecular Pathogenesis
The molecular pathogenesis of glucagonomas is still largely unknown. Glucagonomas may be associated with the MEN-1 characterized by a family history of pituitary, pancreatic islet cell, or parathyroid tumours in 3 % of the cases, and glucagonoma is associated with 13 % of MEN-1-related tumours. The malignant progression of glucagonoma may be associated with complex genetic mechanisms, but no specific molecular markers of malignancy have been defined with certainty so far [8].
15.5 Histopathology
Histologically glucagonomas are classified as well-differentiated functioning pNETs since these tumours demonstrate the typical morphological characteristics of other pNETs. Glucagonomas usually present as encapsulated firm nodules varying in size from several mm’s up to 25 cm (Fig. 15.1) [2, 7]. The majority of glucagonomas are 5–10 cm large in size at the time of diagnosis. The tail of the pancreas is predominantly involved. In one study slightly more than 50 % of glucagonomas occurred in the tail and 80 % in another study. But, in the largest study, 22 % of glucagonomas were in the head region of the pancreas, 14 % in the body, and 51 % in the pancreatic tail. Most glucagonomas are within the pancreas; however, two glucagonomas with the clinical syndrome were found in the proximal duodenum and kidney. Glucagonomas usually occur as a single tumour, although 10–12 % patients have been reported as multicentric tumours or diffuse lesions throughout the pancreas.
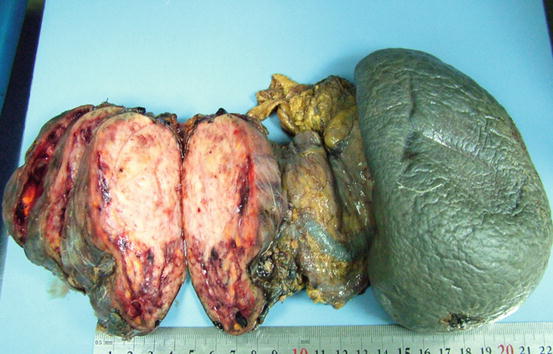
Fig 15.1
Glucagonoma presenting as a firm nodule on macroscopic examination
Immunocytochemical and histologic studies show typical findings of pNETs. The tumours consist of cords and nests of well-differentiated islet cells (Fig. 15.2). Despite their benign histologic appearance and slow growth rate such as low mitotic index and uncommon nuclear atypia, most pancreatic glucagonomas are malignant with high metastatic potential. Sixty percent of glucagonomas were reported as malignant, and 51.4 % of all malignant glucagonomas were metastatic at the time of diagnosis.
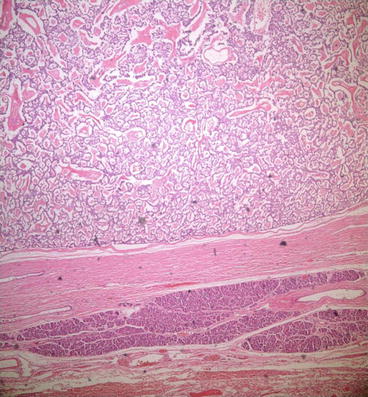
Fig 15.2
Tumor cells forming cords and nests in glucagonoma on microscopic examination
Glucagon is usually detectable within the tumour cells by immunoperoxidase staining and glucagon mRNA may be detected by in situ hybridization. Glucagonomas are often mixed with pancreatic polypeptide-containing cells or less often with insulin- or somatostatin-containing cells. Characteristic alpha cell granules may be seen on electron microscopy. Alpha cell tumours can be divided into two distinct types. Those tumours associated with the glucagonoma syndrome are usually solitary and large, with undescriptive microscopic pattern, atypical granules ultrastructurally, and few cells positive for glucagon immunohistochemically. These ultrastructural and immunohistochemical aberrations may be the result of abnormalities in the biosynthesis and secretion of glucagon, sometimes with alternative production of the related precursor peptides glycentin and oxyntomodulin. The alpha cell tumours which are not associated with the glucagonoma syndrome are often multiple and small, have gyriform pattern of growth, are strongly immunoreactive for glucagon, exhibit typical α cell granules ultrastructurally, and are nearly always benign. In one series of 1,366 autopsy cases, the frequency of adenomas was reported to be 0.8 %, and all contained glucagon-producing cells and argyrophilia can be demonstrated with Grimelius technique [7, 9–12]. In contrast, detection of atypical granules is typical in patients with tumours associated with hyperglucagonemia syndrome.
Skin biopsies are not diagnostic in patients with glucagonoma. The histopathology of NME changes can be numerous as much as the clinical presentation. Typical histological findings include epidermal necrosis, parakeratotic hyperkeratosis, irregular epidermal hyperplasia, papillary dermal angiodysplasia, subcorneal pustules, and suppurative folliculitis. None of these findings indicate a pathognomonic finding; in contrast histologically and clinically, it is similar to the rash of zinc deficiency, acrodermatitis enteropathica. In its classic form, early lesions demonstrate superficial spongiosis or necrosis with subcorneal and midepidermal bullae. Fusiform keratinocytes with pycnotic nuclei are often present with mononuclear inflammatory infiltrates. This characteristic histologic pattern is best seen in early lesions. The most specific feature on skin histological examination is necrolysis of the upper epidermis with vacuolated keratinocytes, leading to focal or confluent necrosis, but this histopathologic feature may be seen in other deficiency states like pellagra, necrolytic acral erythema or zinc deficiency [9].
15.6 Pathophysiology
The etiology and pathogenesis of clinicopathologic characteristics of glucagonoma have not been enlightened yet. But the pathophysiology of glucagonoma syndrome is probably related to the known actions of glucagon. Glucagon stimulates glycogenolysis, gluconeogenesis, ketogenesis, lipolysis, and insulin secretion; alters intestinal secretion and inhibits pancreatic and gastric secretion and gut motility. Hyperglycemia in the glucagonoma syndrome results from the increased glycogenolysis and gluconeogenesis. Because glucagon increases secretion of insulin, which prevents lipolysis and maintains normal free fatty acid concentrations, ketonemia usually does not develop.
Venous thromboembolism, NME, and angular cheilitis were observed in patients treated with continuous intravenous glucagon for refractory tumour-induced hypoglycemia. This observation indicates that glucagon may have a direct, causative role in diabetico-dermatogenic syndrome (DDS) [13]. Normalization of glucagon concentrations by surgery results in a rapid disappearance of the skin rash. Further evidence for the role of preproglucagon-derived peptides in the pathogenesis of necrolytic migratory erythema comes from its rare association with other diseases, notably hepatic cirrhosis and celiac disease. The patient with celiac disease had marked elevation of circulating enteroglucagon levels. In those patients with cirrhosis, glucagon values were not invariably elevated, but hepatocellular dysfunction may result in increased levels of other preproglucagon-derived peptides, such as enteroglucagon. In one case of cirrhosis, the rash was successfully treated with fatty acid and zinc supplementation. However, it is not clearly established that the skin rash is associated with hyperglucagonemia since numerous patients that have been given large doses of glucagon for long periods did not develop skin rash. Hypoaminoacidemia, nutritional lack of zinc and fatty acids, and hepatocellular dysfunction are all considered as possible triggering factors of NME. Hyperglucagonemia provokes multiple nutrient and vitamin B deficiencies, which in turn are the probable cause of this typical skin disorder [14].
15.7 Clinical Features
Skin rash and mild diabetes are the predominant signs of the glucagonoma syndrome. The incidence of this DDS was reported to be as 57.2 % [14]. Glucagonomas predominantly metastasize to the liver and adjacent lymph nodes, less frequently to the vertebra, ovary, peritoneum, and adrenals. Unlike carcinoid syndrome, liver metastases are not a prerequisite for the clinical syndrome to occur. The rash may occasionally appear prior to the onset of systemic symptoms. Most patients with rash usually also have weight loss, diarrhea, sore mouth, weakness, mental status changes, or diabetes mellitus.
The characteristic feature of the glucagonoma syndrome is the rash, which is called as NME. It occurs in 90 % of the cases and is usually the presenting feature. The rash exhibits a 7- to 14-day sequence of erythema, blister, secondary breakdown, infection, healing, and hyperpigmentation. The initial manifestation of the rash is well-demarcated symmetrical areas of erythema (an annular or arciform erythema) at intertriginous and periorificial sites, usually in groins, which migrates principally to the limbs, buttocks, and perineum. These lesions may become vesicopustular or bullous and coalesce, and eroding and encrusting may occur. The rash may be mildly pruritic and occasionally painful. The extent and severity of lesions may wax and wane. The healing process commences after 10–15 days; lesions clear centripetally and leave indurated and hyperpigmented areas, which are usually permanent. The rash remits and relapses unpredictably, and this has made it difficult to assess treatment efficacy. On the basis of this morphology, differential diagnosis includes pemphigus foliaceus, pemphigoid, vasculitis, psoriasis, herpes, seborrheic or contact dermatitis, eczema, pellagra, zinc deficiency (inherited, acrodermatitis enteropathica, or acquired), extensive candidiasis, or even chemical burn. Usually NME rapidly disappears after successful surgical removal of the glucagon-secreting tumour. Although this eruption is a very specific cutaneous marker for underlying glucagonoma, in a few affected patients, no evidence of associated malignant disease has been found. Glucagon itself may act directly on the skin to cause the rash, perhaps by increasing arachidonic acid levels, which it does in human keratinocyte cultures. The rapid response of the rash to octreotide, before changes in circulating amino acids or glucagon, suggests a direct action on the skin, consistent with the inhibition of hormone action. Topical application of zinc or oral supplementation has been reported to improve the rash, although circulating zinc levels are normal in patients with glucagonoma.
In addition to skin rash, the disease may affect the mucous membranes. Angular stomatitis, cheilitis, and atrophic glossitis with a beefy red tongue occur almost invariably. Blepharitis, vulvovaginitis, and urethritis accompanied by dysuria are frequent features. Mucosal lesions are associated with the cutaneous rash in about 70 % of patients. Glossitis or angular stomatitis was reported to occur in 34–68 % of patients. Nail and scalp involvement may result in onycholysis and alopecia. Some patients develop nail dystrophy with brittleness and crumbling.
Amino acid levels are frequently diminished in the serum of patients with glucagonoma. Glucagon, acting on the liver, increases both amino acid oxidation and gluconeogenesis from amino acid substrates [11]. The intensity of hypoaminoacidemia may vary with the intensity of the disease. Glucagon stimulates sustained gluconeogenesis, which depletes the glycogenic amino acids, particularly alanine, serine, and glycine, and hepatic proteolysis with conversion of amino acid nitrogen to urea nitrogen, which depletes all amino acids. It has been postulated that this results in an increase in protein degradation, depleting epidermal protein and ultimately resulting in skin necrosis. Amino acid deficiency is a frequent finding in the glucagonoma syndrome; in two large series, incidence was reported as 26 and 60 %. Plasma concentrations of amino acids are frequently less than 25 % of normal, with glycogenic amino acids most affected, whereas branch-chain amino acids are reportedly less affected. Hypoaminoacidemia may be clinically important because it may be the cause of NME; in one patient, amino acid infusion led to an improvement in the characteristic rash [15]. Intravenous amino acid infusion, although not correcting hypoaminoacidemia, has been reported to ameliorate the rash, but oral amino acid supplementation is ineffective. Similarly a high-protein diet, despite normalizing plasma amino acids and nitrogen balance, may have no effect on the rash. If glucagonoma-induced hypoaminoacidemia is corrected, the dermatitis may improve without changing plasma glucagon levels. The occurrence of the rash in patients without suppressed amino acid levels casts further doubt on a casual association. Resolution of rash, however, has been reported after simple hydration with glucose and saline; therefore, it is not established that either hypoaminoacidemia or zinc deficiency is causative in all patients [7, 9–11].
Glucose intolerance is usually mild and nonketotic since beta cell function is preserved and insulin secretion is normal. Diabetes mellitus occurs in 75–95 % of patients with glucagonoma [16]. Diabetes usually precedes skin rash (and diagnosis) by years, in one study with an average time of 5 years. The hyperglycemia due to glucagonoma may be mild or moderate (median-glycated hemoglobin level of 9.8 % in one series [16]) and is easily controlled by diet, oral agents, or insulin. In another report, 42 % of patients required oral hyperglycemic agents and 24 % insulin. There is little correlation between the plasma glucagon levels and the degree of glucose intolerance. This probably reflects variability in beta (β) cell insulin secretion, which may be influenced by loss of β cells as a result of tumour growth. Other factors may include insulin gene expression in glucagonoma cells, secretion of biologically inactive forms of immunoreactive glucagon by the tumour, depletion of glycogen stores, and downregulation of glucagon receptors. Various treatments have resulted in changes in plasma glucagon concentrations that did not correlate with subsequent changes in blood glucose levels. In cases where infusion of a somatostatin analog decreased plasma glucagon levels in patients with glucagonoma, no effect on plasma glucose level was observed. Furthermore, tumour resection and normalization of blood glucagon may not result in normalization of the glucose tolerance in all of the patients. In some cases removal of glucagonoma improved glucose tolerance. It has been suggested that development of diabetes or glucose intolerance depends to a large degree on the patient’s insulin reserve. If the latter is intact, enhanced insulin release may compensate for the hyperglucagonemia and increased hepatic glucose production. Otherwise, diabetes mellitus or glucose intolerance may develop.
Unrelenting weight loss affects the majority of cases (56–96 % of patients), usually despite a good appetite. Weight loss may be profound. A number of observations suggest that the weight loss is a unique aspect of the syndrome. It is seen even in patients with small tumour without metastatic spread. Weight loss is prominent in patients with small tumour as well as in those with metastatic tumour, suggesting that cachexia is a consequence of the catabolic actions of glucagon. This is supported by a study in which a patient’s weight loss was reversed after being given long-acting somatostatin analog, octreotide, which reduced the plasma glucagon level to a near-normal range. The main cause is probably the organ protein catabolism, which occurs in response to the depletion of amino acids. If the weight loss exceeds 50 % of body weight, it can be fatal. It is not seen early in other pancreatic endocrine tumours, unless malabsorption is present. It is suggested that the anorectic effects of glucagonoma are due to the production of a novel substance by the tumour and not due to glucagon per se because other transplanted glucagonomas producing similar levels of plasma glucagon elevation do not cause anorexia.
Nonspecific abdominal pain has been reported in approximately 12 % of patients. The elevation of preproglucagon-derived peptides would be anticipated to affect gastrointestinal function. Enteroglucagon has been proposed as a humoral mediator of intestinal adaptation in response to injury. It appears to have a trophic action on the intestinal mucosa and elevated levels causing villous hypertrophy. Three patients have been described with an enteroglucagonoma, in whom the principal clinical finding was giant villous hypertrophy. Large-molecular-weight forms of glucagon inhibit gastrointestinal smooth muscle, resulting in constipation. Diarrhea is reported in 14–15 % of patients, occasionally with severe steatorrhea, and occurs far more common than constipation (15 % vs 4 % in one study). The etiology of the diarrhea remains unclear. Jejunal biopsies have been reported to be normal or it may show hypertrophic folds. It remains possible that other hormones secreted by the tumour could be contributing to the development of diarrhea. Rarely, nausea and vomiting occurs, possibly as a result of local tumour effects.
A normochromic-normocytic anemia is common, being present in up to 90 % of patients in some series. It is probably due to the anemia of chronic disease, but a direct effect of glucagon on erythropoiesis has also been suggested [17]. The anemia is usually mild with a mean hemoglobin concentration of 9.4 g/dl, but occasionally the hemoglobin may fall as low as 4 g/dl. Serum iron, B12, and serum folate concentrations usually are normal, and anemia does not respond to therapy with any of these agents. It does respond to resection of the tumour, however. Further evidence of anemia related to glucagon excess is that the prolonged therapy with a long-acting glucagon preparation decreased erythropoiesis in rats and mice.
Related posts:
 Molecular Biology of Neuroendocrine Tumors
Introduction to Neuroendocrine Tumours
Somatostatin Analogs and Interferon in the Treatment of Neuroendocrine Tumors
Molecular Biology of Neuroendocrine Tumors
Introduction to Neuroendocrine Tumours
Somatostatin Analogs and Interferon in the Treatment of Neuroendocrine Tumors
 Liver-Directed Therapies in Neuroendocrine Tumors
Liver-Directed Therapies in Neuroendocrine Tumors
 Thyroid Cancer
Thyroid Cancer
 Pathological Evaluation and Classification of Digestive Neuroendocrine Tumours
Pathological Evaluation and Classification of Digestive Neuroendocrine Tumours
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


